Neurofibroma
EPIDEMIOLOGY
Neurofibromas often occur as solitary lesions and are common in the general population. NF1 has a prevalence of 1 in 4000.
ETIOLOGY AND PATHOGENESIS
Neurofibromas are hyperplasias of all the nerves elements. Neurofibromas can be seen as sporadic lesions or in the context of NF1,. The gene for NF1 is located on the long arm of chromosome 17 (17q11.2). At least one of the functions of the NF1 protein, neurofibromin, is regulation of the ras oncogene. Although it has been shown that ablating NF1 in mice can lead to the development of neurofibromas, the effects on the levels of ras are not consistent and, thus, other stromal or genetic effects must contribute to the development of neurofibromas.
CLINICAL FINDINGS
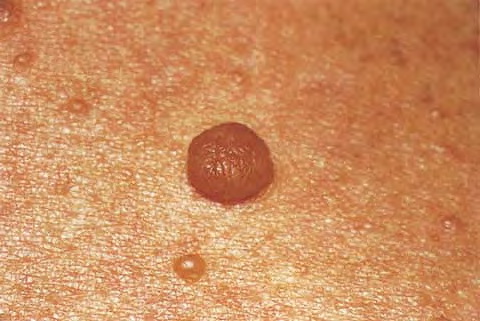
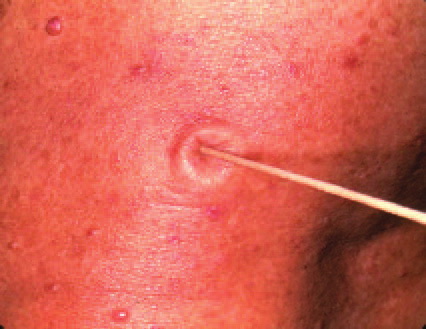
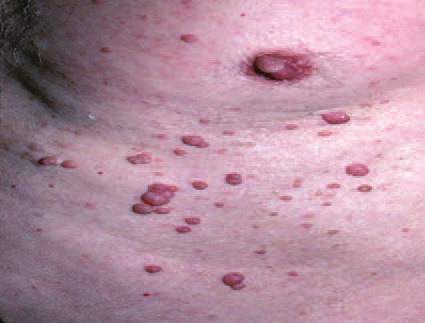
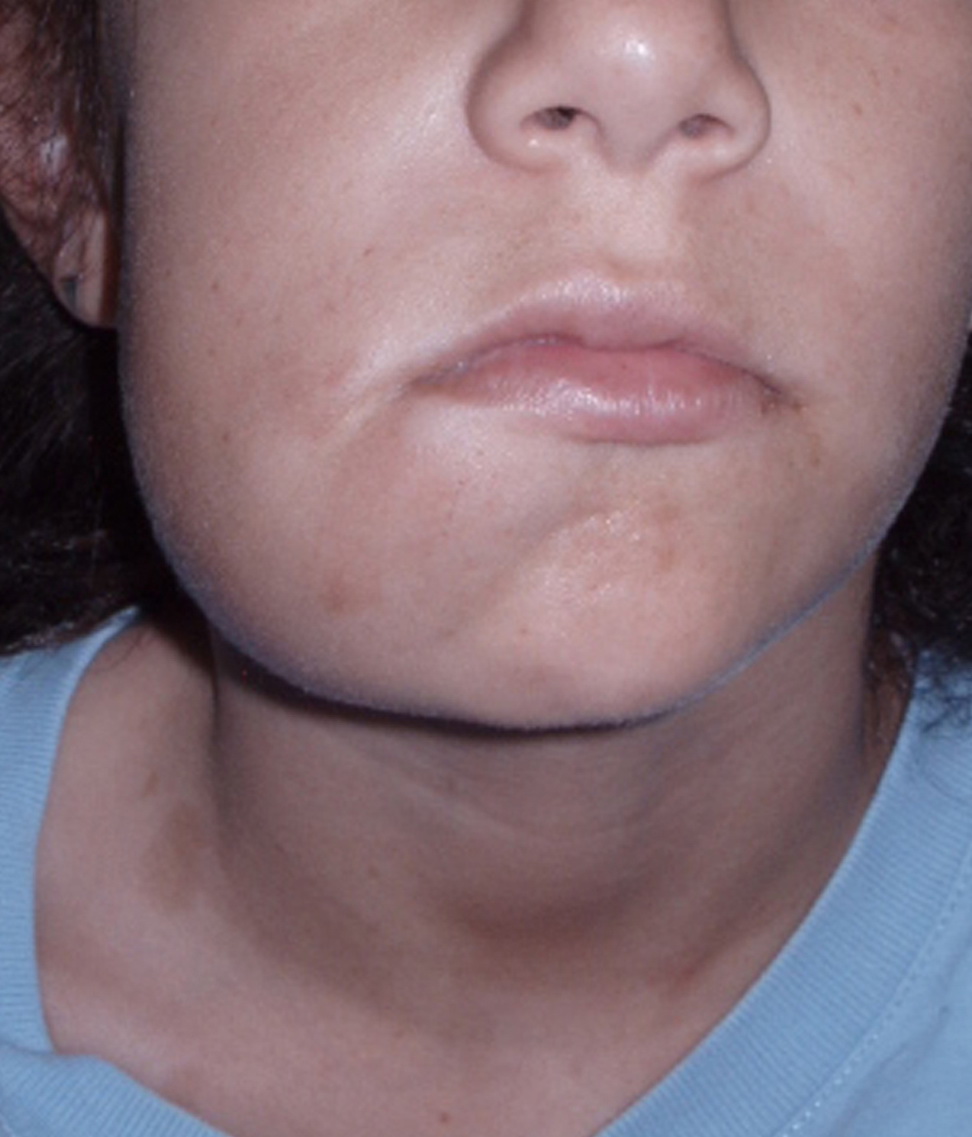
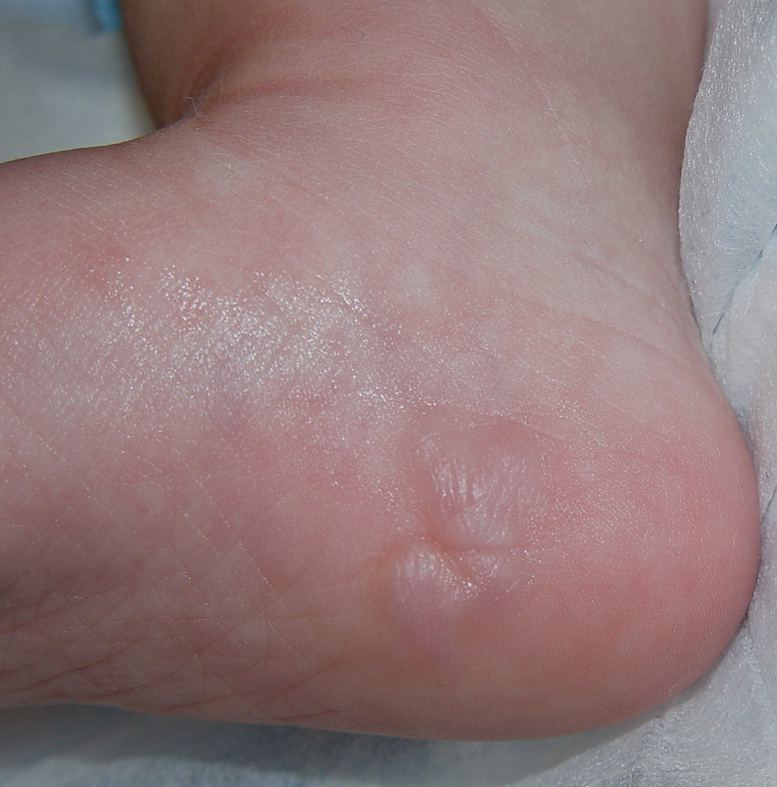
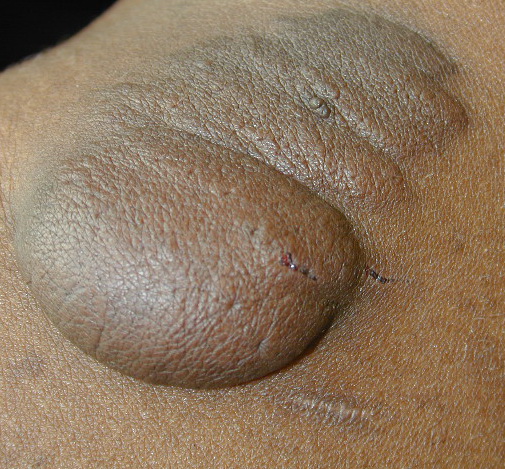
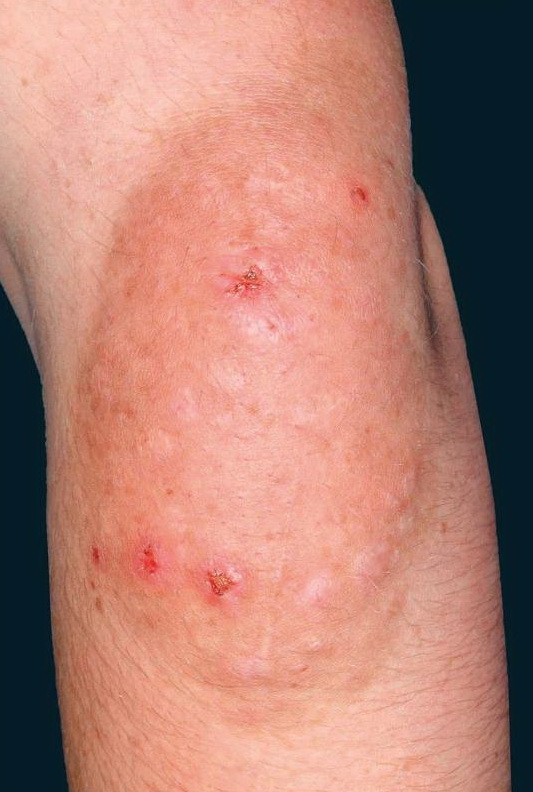
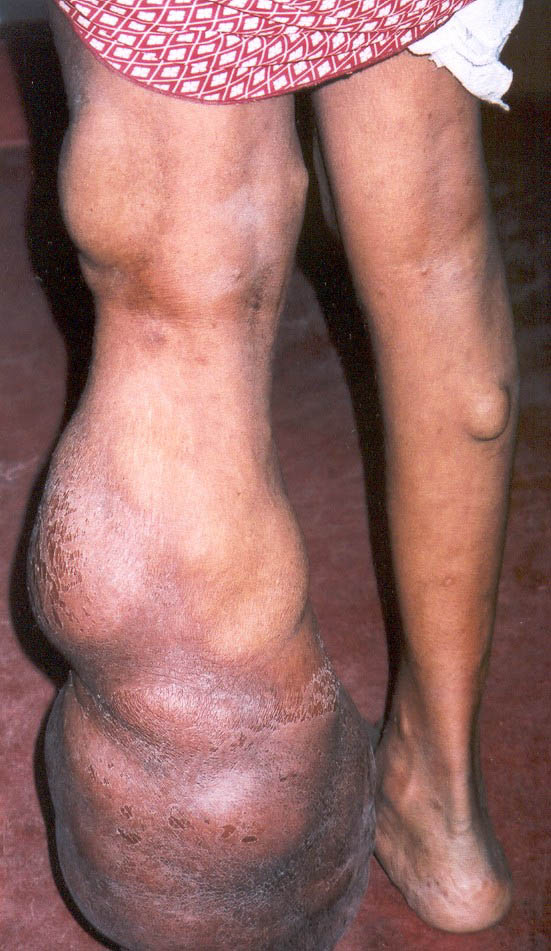
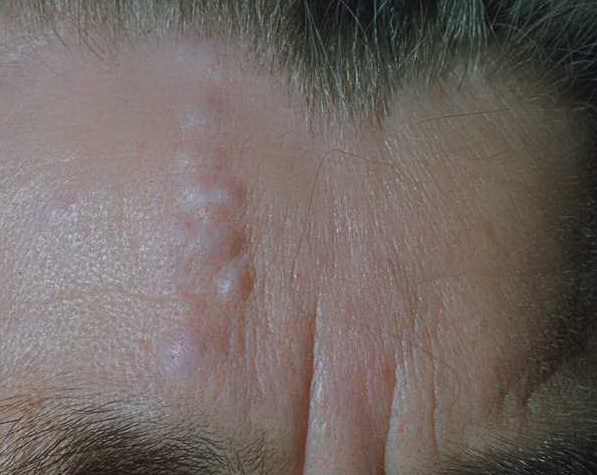
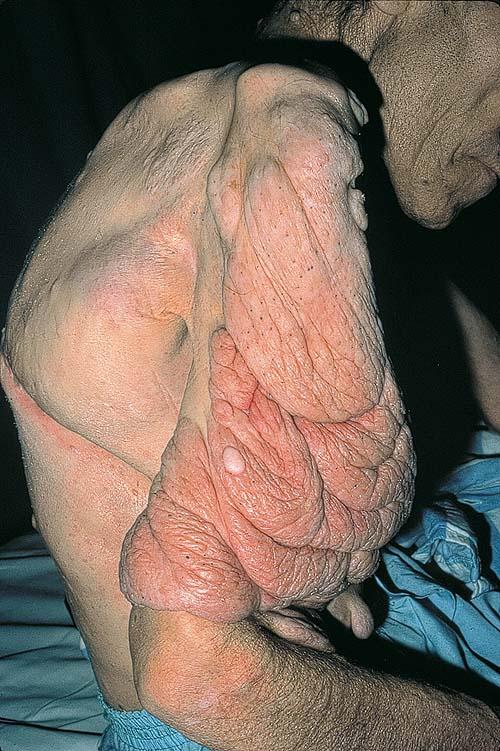
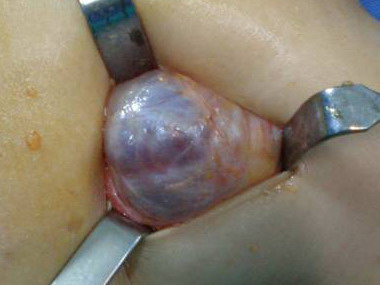
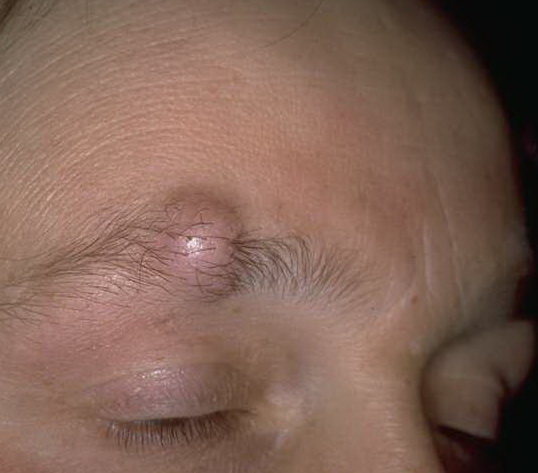
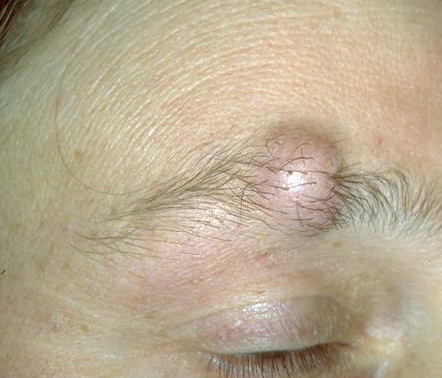
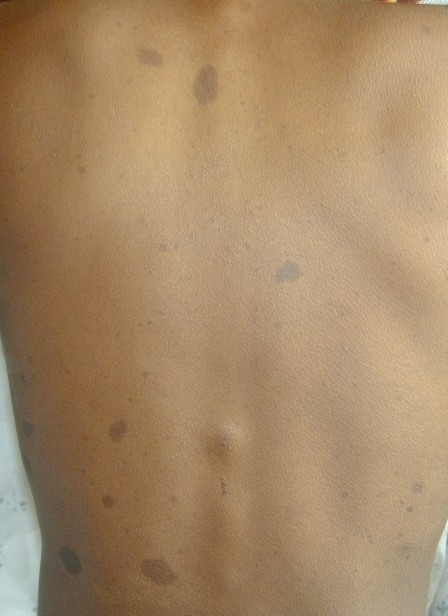
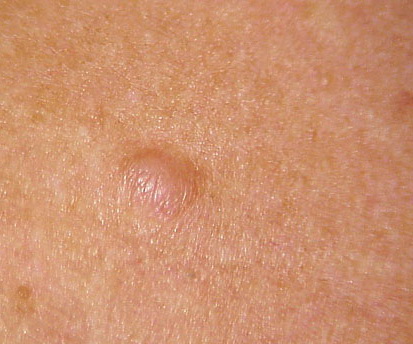
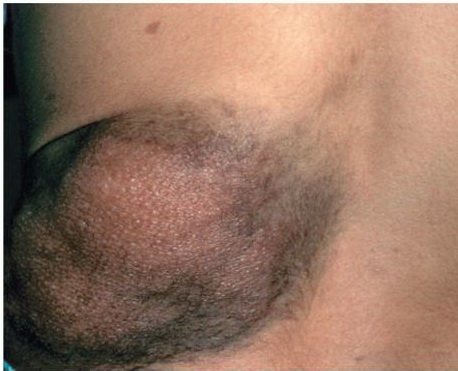
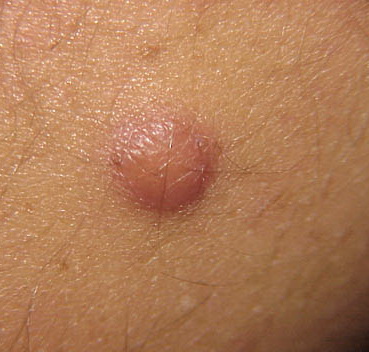
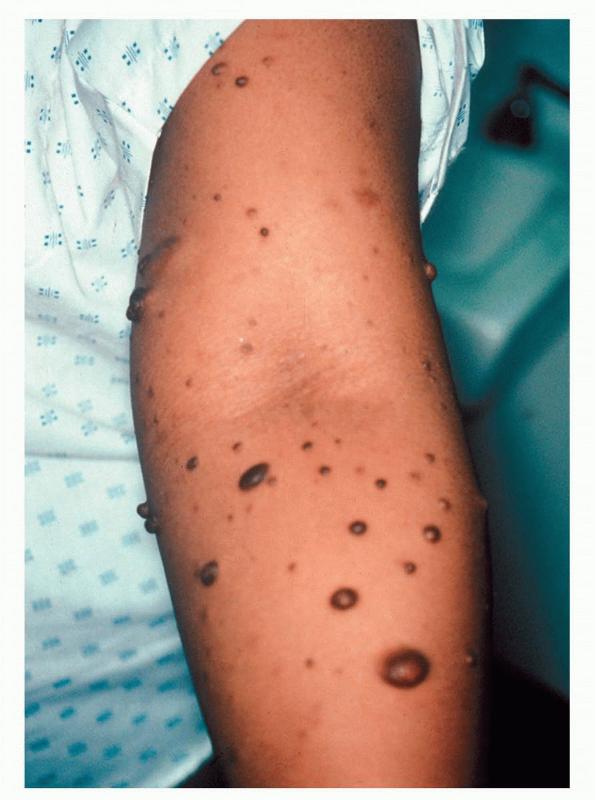
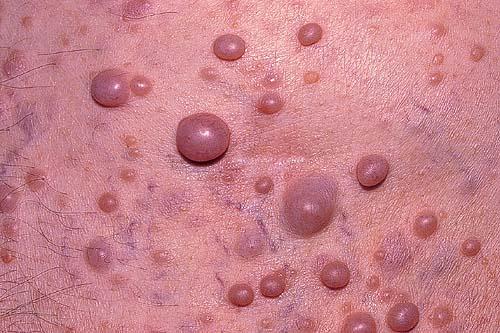
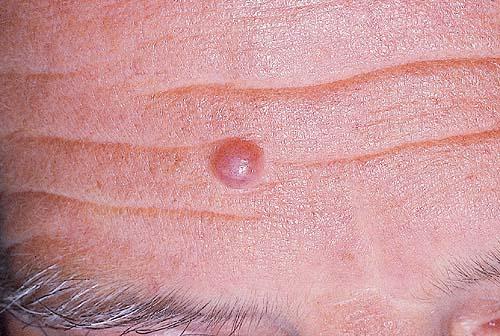
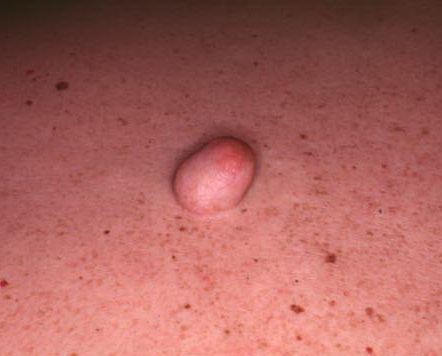
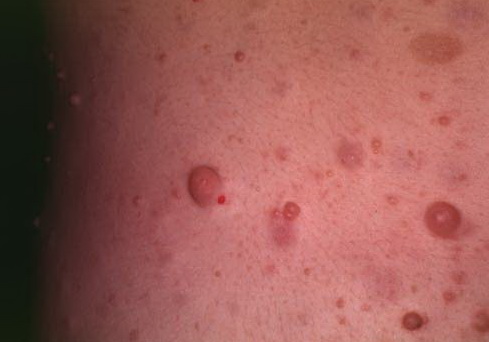
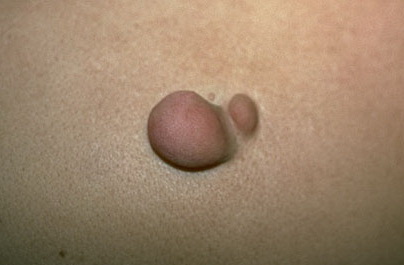
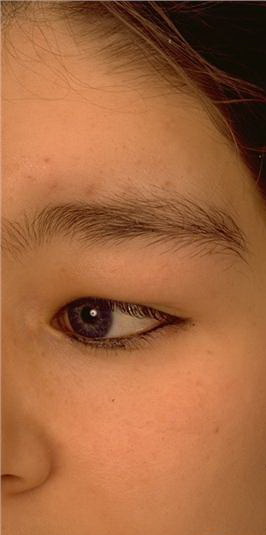
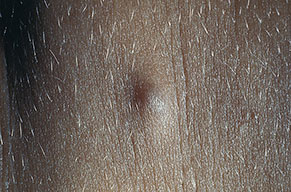
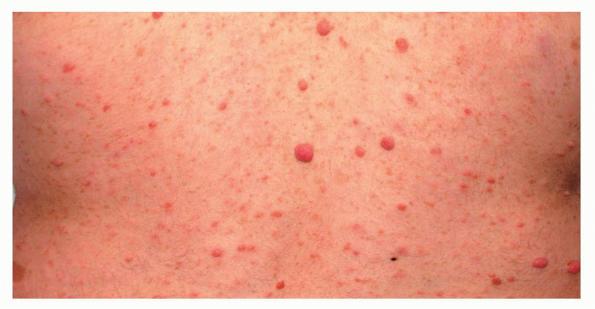
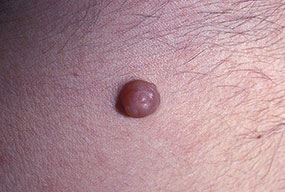
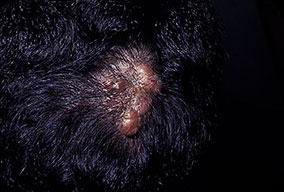
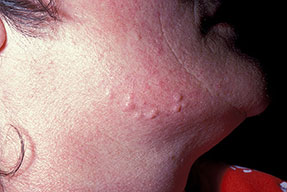
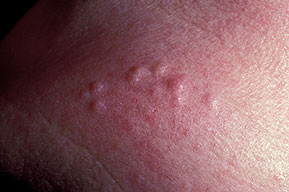
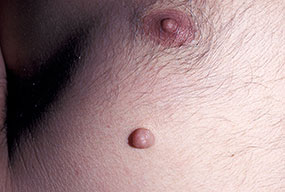
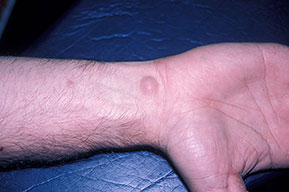
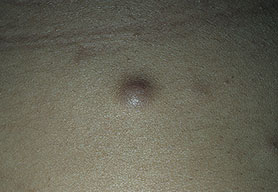
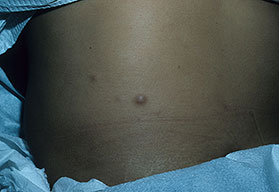
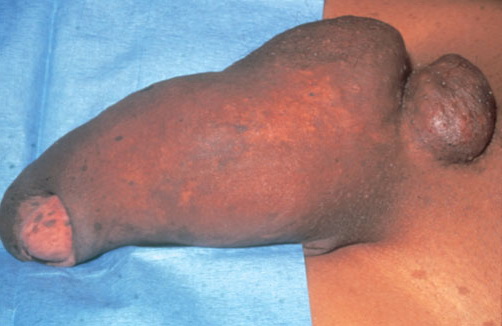
Cutaneous neurofibromas present as protuberant to pedunculated, flesh-colored, soft papules or nodules. They are usually asymptomatic, but they can be pruritic. Subcutaneous neurofibromas are usually larger than dermal lesions and consist of a fusiform swelling involving a larger nerve. In patients with NF1, the tumors start appearing in early childhood and may be quite variable in size. NF1 has a highly variable expressivity, even among patients from the same kindred who have identical germline mutations. Another type of neurofibroma is the plexiform variant , which involves an entire large nerve and its branches (see eFig. , forming a mass of tangled, rope-like structures that feel similar to a “bag of worms” on palpation and can be associated with massive soft-tissue overgrowth, leading to functional impairment.
HISTOPATHOLOGY
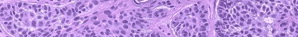
Histologically, a neurofibroma is composed of Schwann cells, fibroblasts, endothelial cells, perineurial fibroblasts, mast cells, and axons, all arranged haphazardly in a matrix that contains collagen and myxoid ground substance in various proportions . Dermal neurofibromas are circumscribed but unencapsulated nodules. A rare sub-type of neurofibroma is the diffuse variant that involves the skin and subcutaneous tissue in a plaque-like fashion, surrounding rather than displacing pre-existing structures such as skin appendages. Plexiform neurofibromas have the same histologic appearance as ordinary neurofibromas but involve an entire nerve and its branches; they also infiltrate the surrounding soft tissue. A variant with a very unusual histologic appearance has been described as dendritic cell neurofibroma with pseudorosettes.72 Finally, some neurofibromas are melanotic.
DIFFERENTIAL DIAGNOSIS
Differential diagnosis of neurofibroma includes fibrolipoma, lipoma, and MPNST.
TREATMENT
Solitary dermal neurofibromas can be excised. Management of patients with NF1 andis discussed in Chap. 142. Resection is currently the best treatment option for plexiform neurofibromas; however, the lesions frequently recur after resection. Recurrence is more commonly associated with a young patient age, incomplete tumor resection, or non-extremity tumor location