
Contact Us :
👤 Users: 0 | 👁 Guests: 2 | 🌍 Total: 2
Erythrokeratoderma variabilis=تقرن الجلد الاحمراري المتباين


Erythrokeratodermia variabilis
Erythrokeratodermia variabilis (EKV) is a rare genetic skin disorder listed in Online Mendelian Inheritance in Man (OMIM) # 133200. Erythrokeratodermia variabilis belongs to the clinically and genetically heterogeneous group of erythrokeratodermas.
Erythrokeratodermia variabilis is characterized by the coexistence of 2 distinct morphologic features: hyperkeratosis and transient erythema. de Buy Wenninger recognized and described the first cases of erythrokeratodermia variabilis in the Netherlands in 1907.1 In 1925, Mendes da Costa presented a detailed clinical description of the disease in a mother and daughter, reviewed 8 similar cases that were previously published, and coined the name “erythro- et keratodermia variabilis.”2 During the next decades, multiple case reports emerged in the Northern European literature, including a study of 33 affected members of a Dutch family and 29 affected persons in 5 generations of a Swiss family.3 In 1964, Barsky and Bernstein reported the first case in the American literature.4
Pathophysiology
Erythrokeratodermia variabilis is an inherited disorder of cornification associated with noninflammatory erythema. Marked hyperkeratosis is present, probably because of an increased proliferation and disturbed differentiation of keratinocytes.
In approximately two thirds of erythrokeratodermia variabilis patients, mutations have been identified in 2 connexin genes, GJB3 encoding connexin-31 and GJB4 encoding connexin-30.3. Connexins are a family of transmembrane proteins that assemble into hexameric hemichannels and form gated intercellular gap junction channels. The finding of mutations in GJB3 and GJB4 suggests that the clinical manifestations of erythrokeratodermia variabilis are caused by impaired gap junctional intercellular communication or hemichannel function due to a dominant effect of mutant gap junction proteins.
History
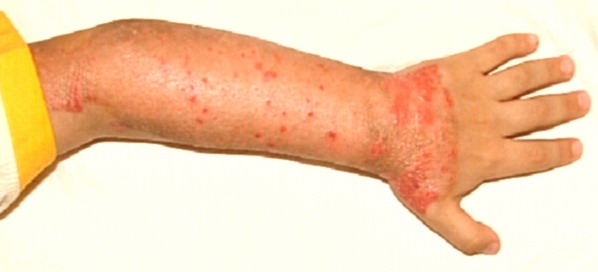
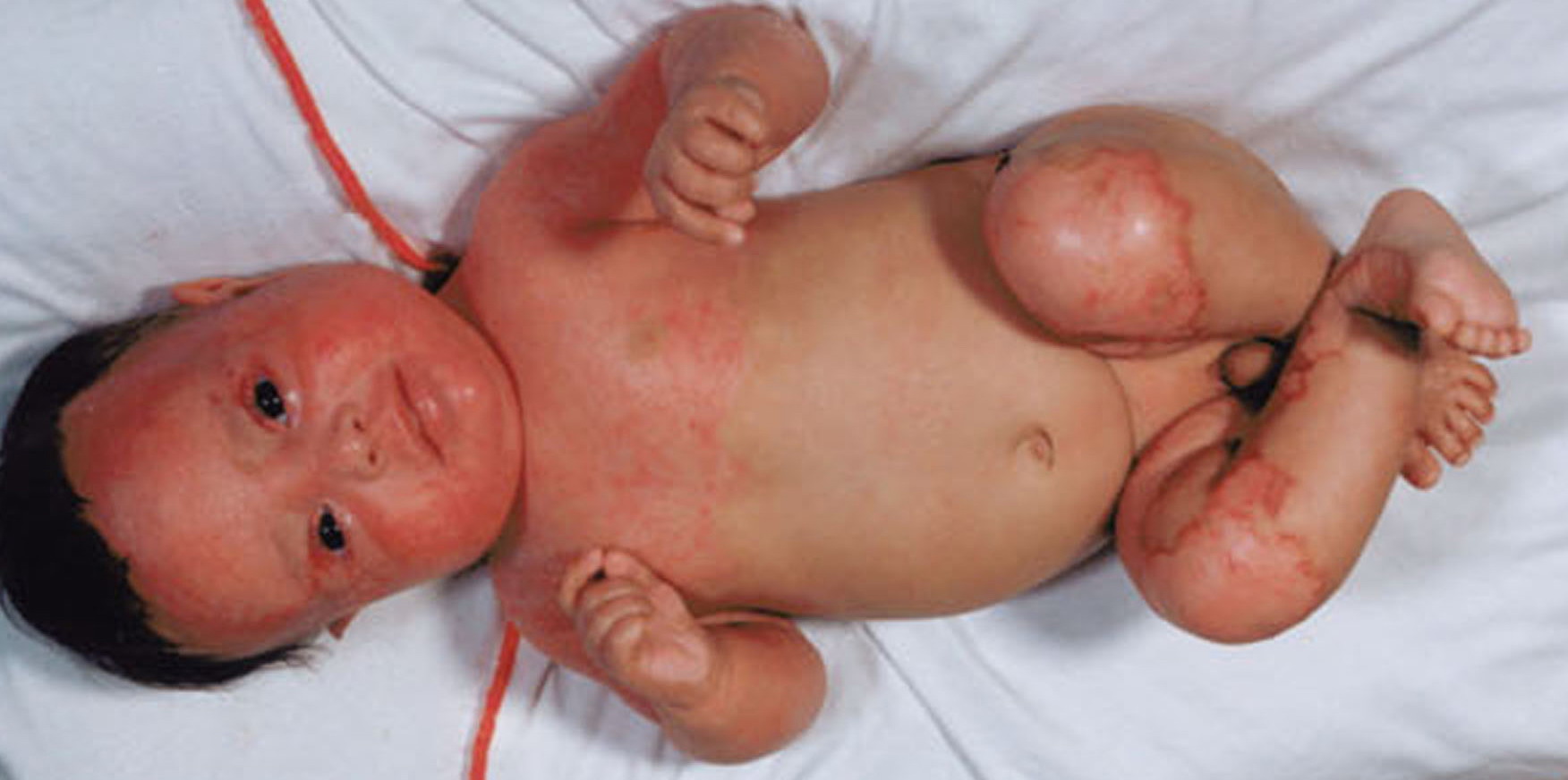
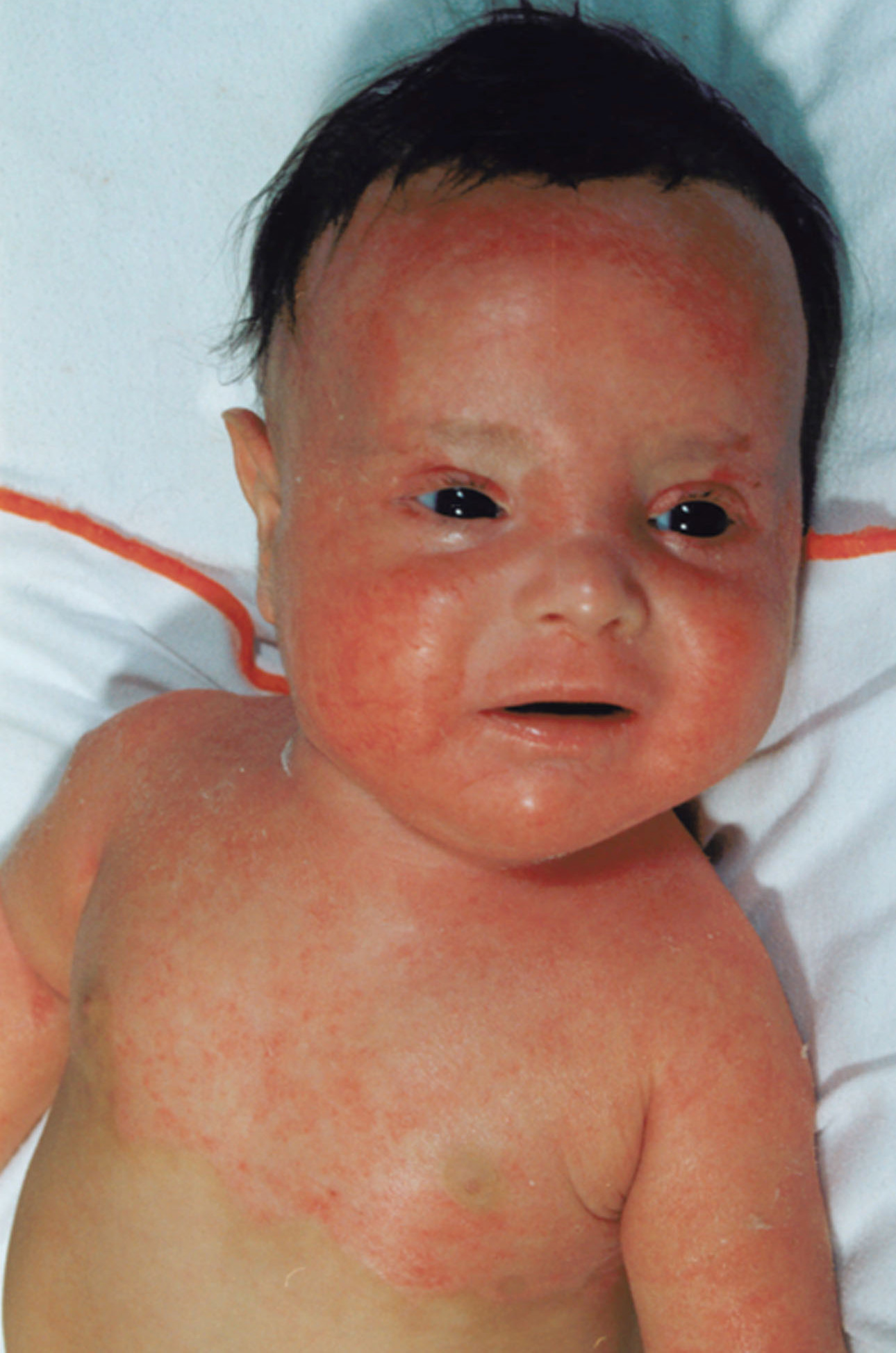
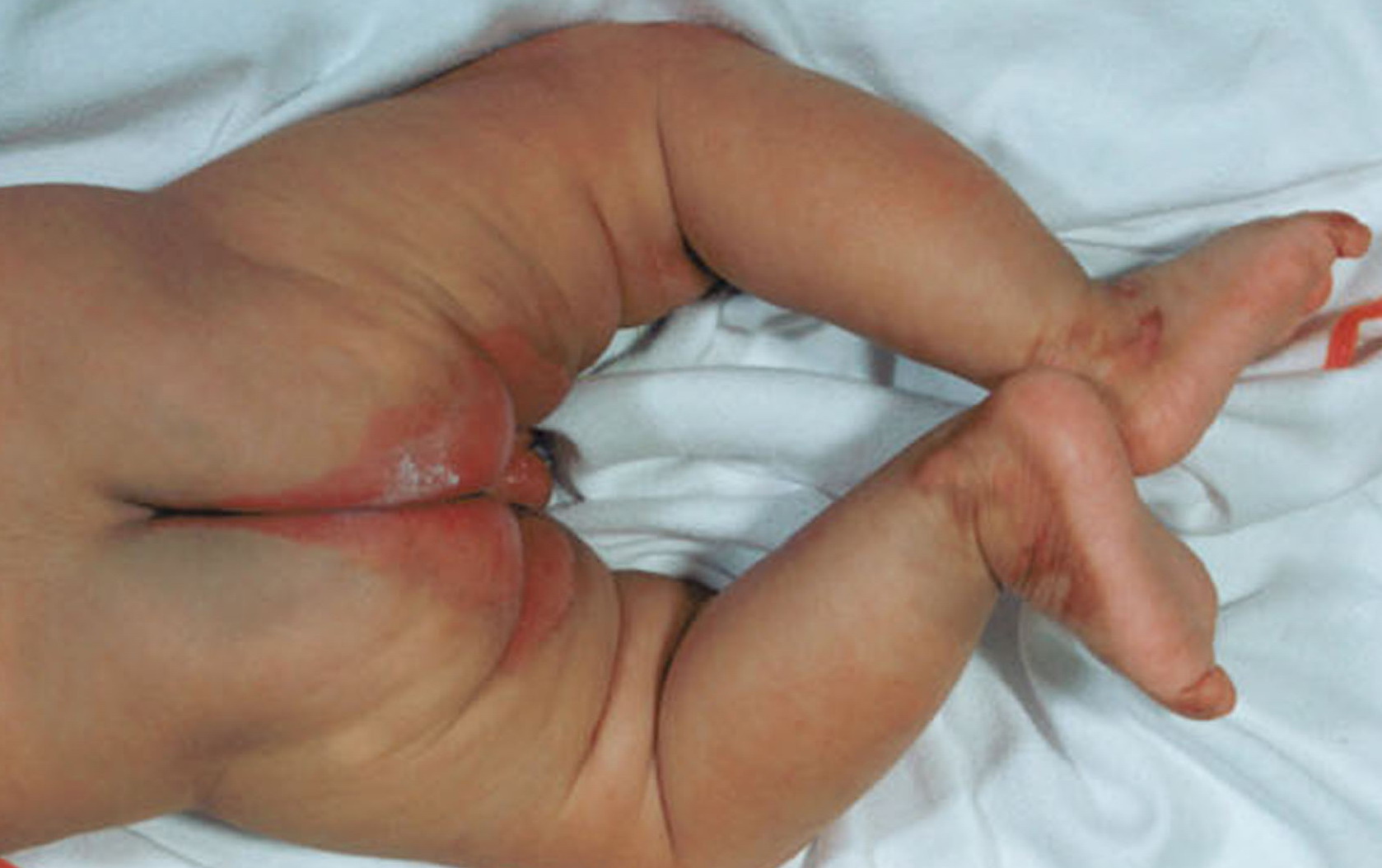
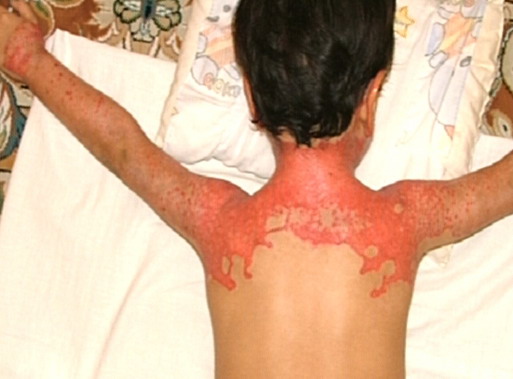
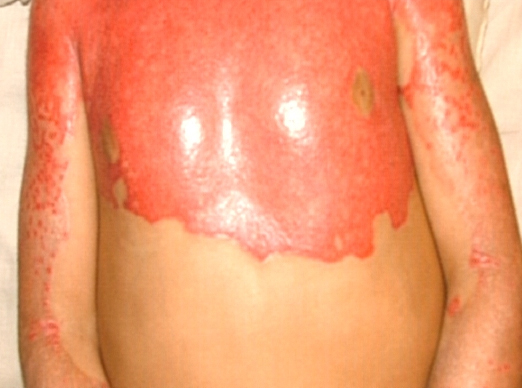
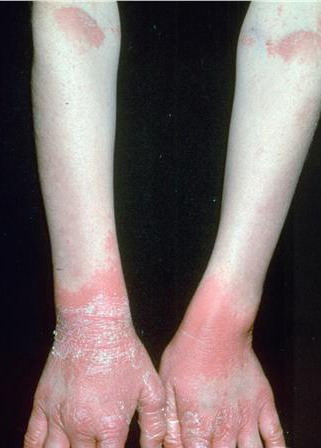
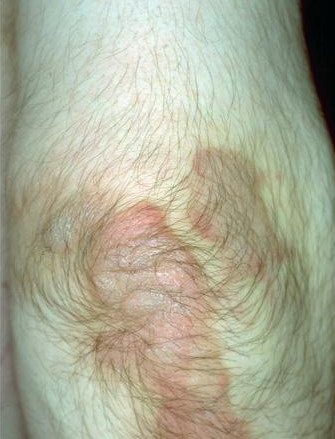
Most erythrokeratodermia variabilis patients initially present with transient, circumscribed, figurate erythematous patches that may involve any part of the integument. These lesions are most prevalent during childhood and may become less frequent as the patient ages.
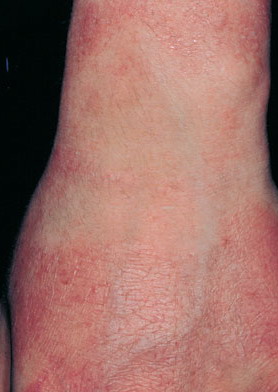
Concurrently or over time, a thickening of the skin (hyperkeratosis) develops, which may be generalized or localized with yellow-brown, thickened, rough, hyperkeratotic plaques on the extremities and trunk. These hyperkeratotic plaques are relatively stable and last for months to years, but they can also clear completely.
After erythrokeratodermia variabilis progresses throughout the patient’s infancy and childhood, it seems to stabilize after puberty and slowly regresses when the patient is older. Improvement and periodic clearing of the skin are not unusual.
Skin lesions may be triggered by internal and/or external factors. These factors include stress, sudden temperature changes, cold, mechanical friction, and, rarely, sun exposure
- Predominance of circinate erythematous patches has been reported in patients with GJB4 (Cx30.3) mutations and has also been described as erythrokeratodermia variabilis with erythema gyratum repens –like lesions.
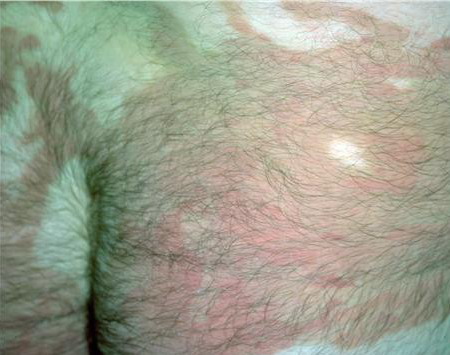
- Erythema can appear on healthy skin and within hyperkeratotic plaques.
- The individual erythematous lesions are transient, usually persisting only for minutes to hours, although they may last for days.
- In about 35% of patients, erythema may be preceded or accompanied by a burning sensation, which may cause serious discomfort for patients.
- The remarkable variability of the erythematous patches in number, size, shape, location, and duration is a typical feature of erythrokeratodermia variabilis that is reflected by the name of the disease.
- Hyperkeratosis
- Hyperkeratosis may be localized or generalized, but tends to be consistent within a family.
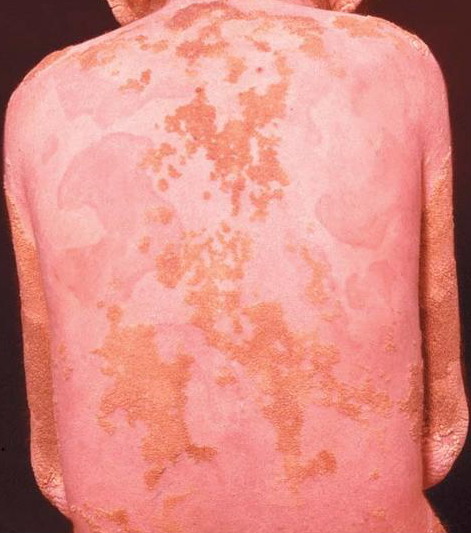
- The generalized form of hyperkeratosis manifests as persistent, yellow-brown-gray thickening of the skin with accentuated skin markings.
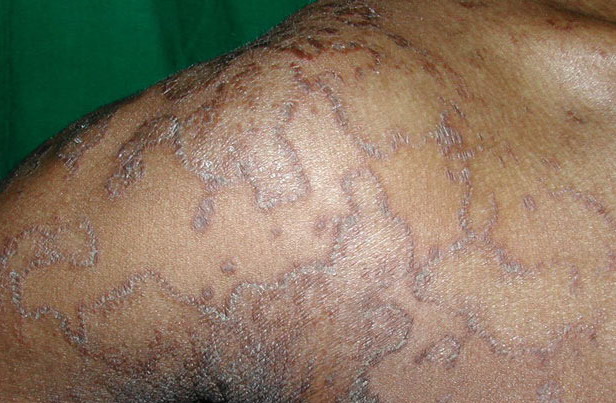
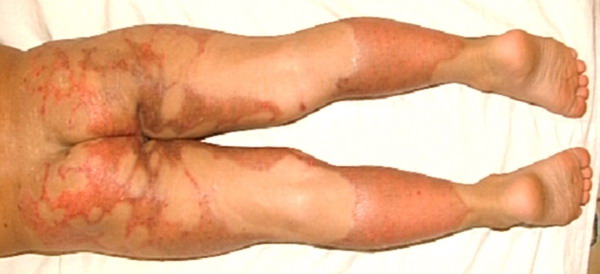
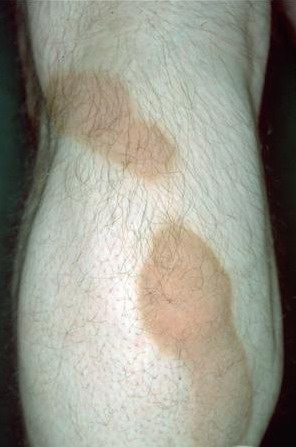
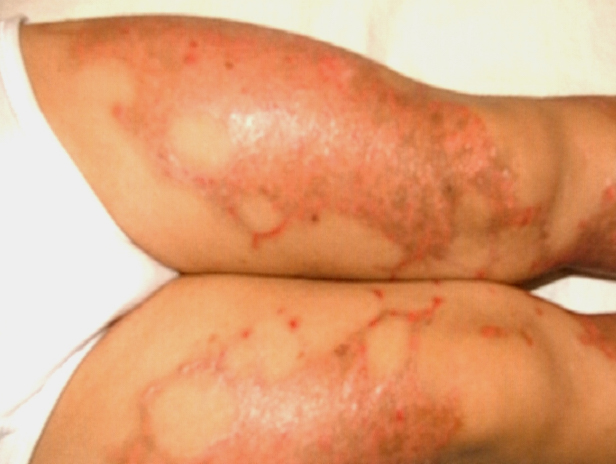
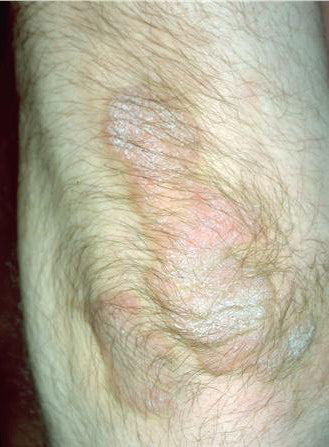
- Rarely, thickened plates of gray-dark brown hyperkeratosis with a spiny, hystrixlike appearance, as in the image below, are present on the lower extremities.
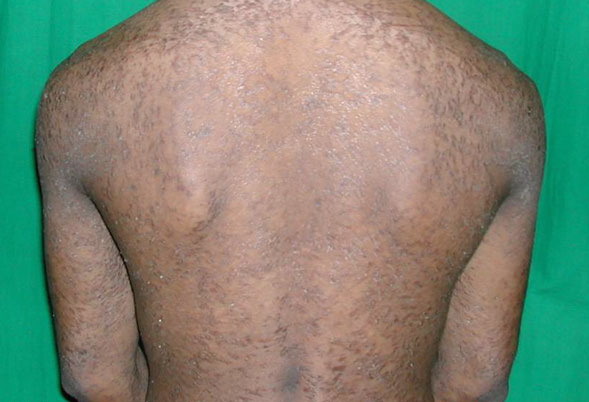
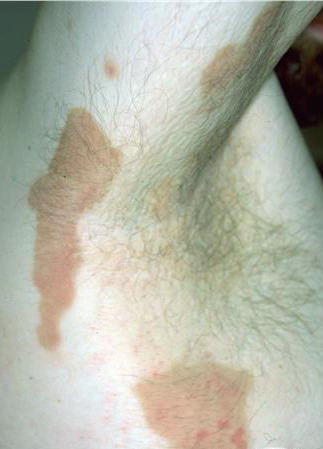
The localized form is characterized by sharply demarcated, brownish, hyperkeratotic plaques with figurate outlined borders.
- Their surface may be ridged and verrucous or show a collarettelike peeling or fine scaling.
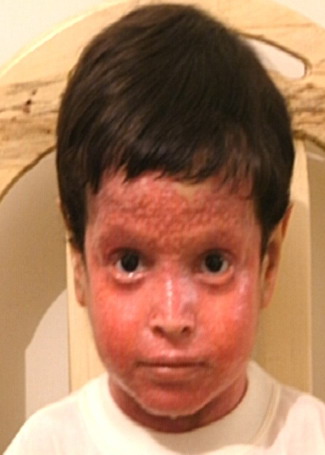
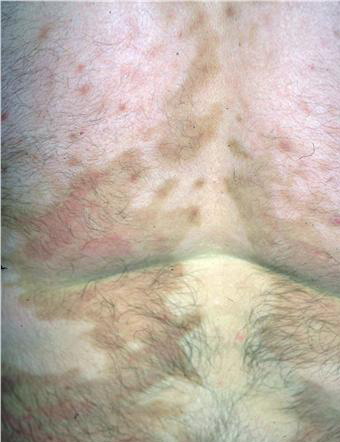
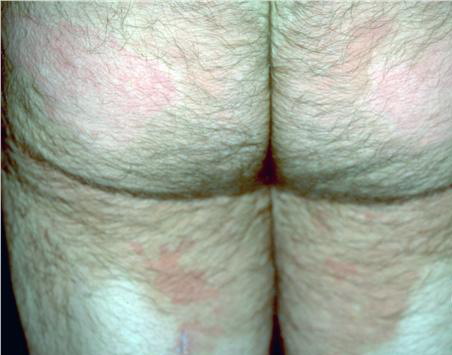
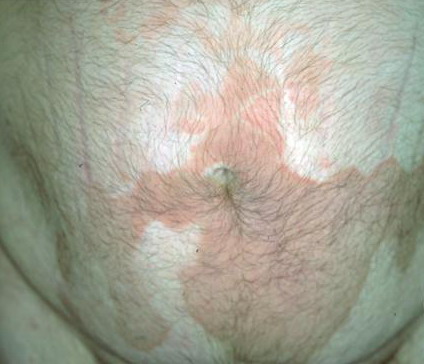
- The plaques are almost symmetrically distributed, as shown below, over the limbs, buttocks, and trunk; often, the flexures, face, and scalp are spared.
- Hyperkeratosis may be localized or generalized, but tends to be consistent within a family.
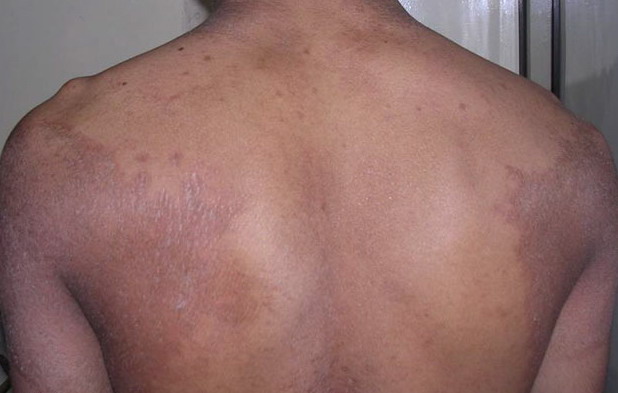
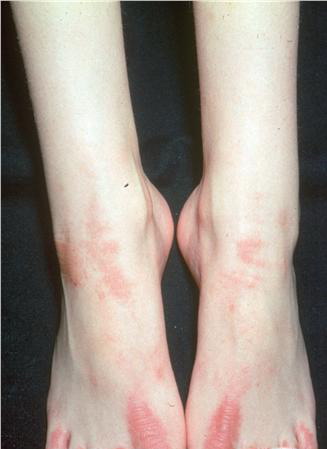
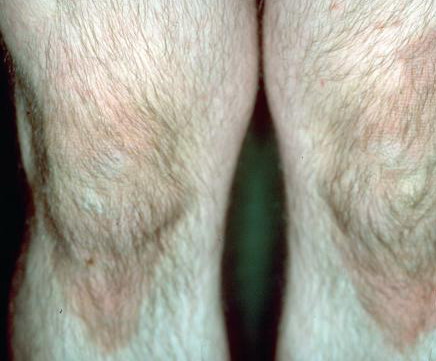
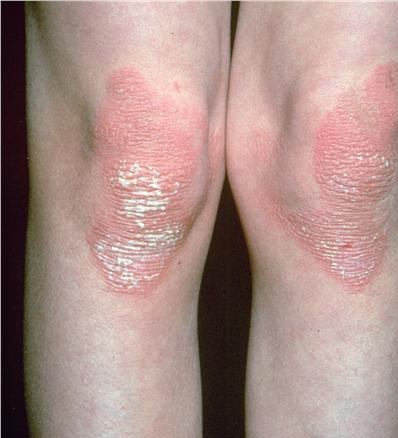
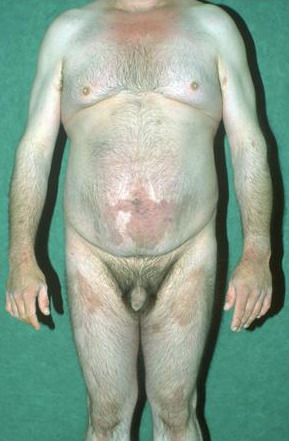
- Relatively fixed lesions over knees, elbows, Achilles tendons, dorsum of the feet, and belt area, as depicted below, are common and can persist for months or years.
- Nevertheless, individual plaques may change size and shape; they may also regress, leaving healthy skin in their place.
- Sometimes, hyperkeratotic plaques have hyperpigmented borders or are associated with hypertrichosis.
- Over the distal joints, the surface of these plaques may become velvety or have a cobblestone pattern.
- In about half the affected families, hyperkeratosis involves the palms and soles of the feet as a patchy or diffuse palmoplantar keratoderma. Often, this palmoplantar hyperkeratosis is associated with peeling
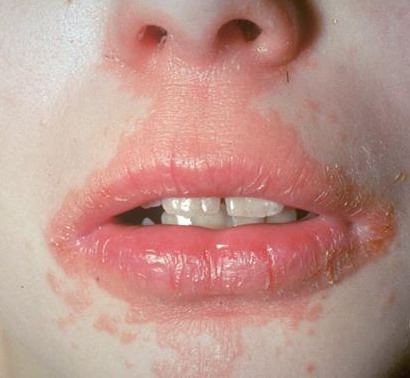
- Hair, nails, teeth, and mucous membranes are not involved.
Causes
Erythrokeratodermia variabilis is usually inherited in an autosomal dominant pattern with nearly complete penetrance. A respectable number of sporadic cases and 2 families with autosomal recessive inheritance have been documented.
The disorder maps to a connexin gene cluster at band 1p34.3.
Erythrokeratodermia variabilis is genetically heterogeneous and caused by mutations in different genes.5,6,7
- Two disease genes have been identified, GJB3 encoding connexin 31 (Cx31) and GJB4 encoding connexin 30.3 (Cx30.3). To date, pathogenic connexin gene mutations have been reported in 24 unrelated erythrokeratodermia variabilis patients and families, including 10 distinct missense mutations in GJB3 and 7 missense mutations in GJB4.8,9,10,11,12,13,14,15
- Several cases of erythrokeratodermia variabilis without identifiable connexin gene mutations have been observed, suggesting that other disease genes exist.16,17
Both Cx31 and Cx30.3 belong to the group of beta-type connexins and are preferentially expressed in the upper, differentiated keratinocytes of human epidermis, suggesting they play a crucial role during epidermal differentiation.18
In vitro expression studies suggest that erythrokeratodermia variabilis mutations disturb the intracellular processing and trafficking of gap junction proteins to the plasma membrane, alter gap junction communication, and induce cell death
Other Problems to Be Considered
Progressive symmetric erythrokeratodermia (PSEK): PSEK is considered an autosomal dominant genodermatosis with a less well-defined clinical presentation than erythrokeratodermia variabilis (EKV). The disease causes fixed, slowly progressive, symmetric, and well-defined hyperkeratotic plaques, which predominantly appear on the extensor surface of the extremities, trunk, and face.
- In contrast to erythrokeratodermia variabilis, no independent, migrating red patches are present, the hyperkeratosis develops on an erythematous base, and the palms and soles are affected more often. The considerable phenotypic variability of PSEK and erythrokeratodermia variabilis and the observation of both phenotypes in one family raised the question of whether PSEK and erythrokeratodermia variabilis are distinct entities or entities within the spectrum of a single disorder. The reported family involved 2 affected children of unaffected parents; one child had the features of erythrokeratodermia variabilis, and the other had the features of PSEK, although the ultrastructural findings in both patients were similar.
- Sequence analysis of the erythrokeratodermia variabilis genes GJB3 (encoding Cx31) and GJB4 (encoding Cx30.3) did not reveal any mutations in 2 families and 4 sporadic cases with the PSEK phenotype, indicating that PSEK and erythrokeratodermia variabilis are distinct clinical entities.
- A frameshift mutation in the loricrin gene (ie, 709insC) on chromosome 1q was identified in a Japanese family with PSEK-like features and mutilating palmoplantar keratoderma (ie, pseudo-ainhum), which is usually not seen in PSEK. This observation is consistent with other loricrin gene mutations in mutilating palmoplantar keratoderma with ichthyosis (Camisa-type of palmoplantar keratoderma; Vohwinkel syndrome with ichthyosis, OMIM 603324) and these disorders are now classified as loricrin keratoderma, an entity distinct from PSEK.20,21 In 2006, a genetic locus for PSEK has been suggested on 21q11.2-q21.2.22
Giroux-Barbeau erythrokeratoderma and ataxia (OMIM 133190)
Greither disease (ie, keratosis palmoplantaris transgrediens et progrediens): This is now known to be a variant of epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma). Two unrelated families have been described with mutations in the keratin 1 (KRT1) gene.
Erythrokeratolysis hiemalis (OMIM 148370, keratolytic winter erythema, Oudtshoorn skin)
Ichthyosis linearis circumflexa (a manifestation of Netherton syndrome)
Annular epidermolytic ichthyosis (as subtype of epidermolytic hyperkeratosis [EHK, bullous ichthyosiform erythroderma])
MEDNIK (mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, and keratodermia) syndrome: Erythema and hyperkeratosis resembling erythrokeratodermia variabilis have been reported in a novel, autosomal recessive genetic syndrome observed in the Bas St-Laurent region of Quebec. Besides skin findings, patients have sensorineural hearing loss, peripheral neuropathy, psychomotor retardation, congenital chronic diarrhea, and an elevation of very long-chain fatty acids. This disorder was mapped to 7q22, and a splice site mutation in the AP1S1 gene encoding the small subunit of the AP1 complex has been identified.23 Originally, this syndrome was reported as “EKV 3 (Kamouraska type),” although that this syndromic disorder is obviously clinically and genetically distinct from erythrokeratodermia variabilis.
Histologic Findings
The histopathologic features in erythrokeratodermia variabilis include orthokeratotic hyperkeratosis, moderate-to-severe acanthosis, and papillomatosis. Although these features are nonspecific, the basket-weave hyperkeratosis may indicate erythrokeratodermia variabilis. In addition, dilated and elongated capillaries are present, with little perivascular inflammation in the papillary dermis. Severe papillomatosis with suprapapillary thinning may result in a church-spire configuration of the epidermis
Medical Care
The goals of the therapy are to diminish the hyperkeratosis and minimize the discomfort.
Systemic retinoids are the treatment of choice in extensive erythrokeratodermia variabilis (EKV).24,25
- Systemic retinoid therapy with acitretin (Soriatane) or isotretinoin (Accutane) can induce dramatic improvement.
- Systemic retinoids are the treatment of choice for extensive or generalized erythrokeratodermia variabilis.
- The effect of acitretin or etretinate is superior to that of isotretinoin.
- The use of retinoids should be considered carefully because long-term therapy is required to achieve continuing results.
- The minimal effective dose for persons with erythrokeratodermia variabilis usually is very low.
The use of topical agents in the management of erythrokeratodermia variabilis depends on the symptoms and focuses on hydration, lubrication, and keratolysis.
- Therapy may include the use of emollients and keratolytics, such as urea, alpha-hydroxy acids, propylene glycol, salicylic acid, and topical vitamin D analogs and retinoid preparations.
- Newer synthetic retinoids, such as short-contact topical tazarotene, combined with moisturizers seem promising.26
- Masking the erythematous lesions of uncovered skin with makeup and other forms of camouflage may provide a cosmetic benefit.
- In case of pruritus and burning, mild sedative antihistamines are useful.
- The avoidance of trauma to the skin may be also beneficial.
Medication
The goals of pharmacotherapy are to reduce morbidity and prevent complications.
Retinoids
Systemic retinoid therapy can induce dramatic improvement. Long-term therapy is required to achieve continuing results. The minimal effective dose for persons with EKV is usually very low.
Acitretin (Soriatane)
Retinoic acid analog similar to etretinate and isotretinoin. Etretinate is the main metabolite; it has demonstrated clinical effects similar to those of etretinate. The mechanism of action is unknown.
Adult
25 or 50 mg/d PO initially; given as single dose with main meal; 25-50 mg/d maintenance; terminate therapy when lesions have resolved sufficiently
Pediatric
Not established
Interferes with contraceptive effect of microdosed minipill progestin contraceptives; not known whether other progestational contraceptives (eg, implants, injectables) are adequate methods of contraception during acitretin therapy; not established if pharmacokinetic interaction occurs between acitretin and other birth control pills
Ethanol causes acitretin to be reesterified to etretinate, which has a much longer half-life; concomitant vitamin A should be limited (package insert says no vitamin A); glibenclamide (a sulfonylurea) potentiated glucose-lowering effect in 3 of 7 patient studied; methotrexate increases risk of hepatitis (do not use concurrently); protein binding of phenytoin may be reduced; tetracyclines increases risk of pseudotumor cerebri (do not use concurrently)Absolute: Pregnancy, likely to become pregnant, or intend to become pregnant within 3 y following cessation of treatment; females who cannot use reliable contraception while undergoing treatment and for at least 3 y after treatment; noncompliance with contraception; breastfeeding; concurrent use of methotrexate (increased liver toxicity) or tetracyclines (pseudotumor cerebri); documented hypersensitivity
Relative: Leukopenia, moderate-to-severe cholesterol or triglyceride elevation; significant Hepatic or renal dysfunctionPregnancy
X – Contraindicated; benefit does not outweigh risk
Precautions
Monitor ALT, AST, cholesterol, triglycerides, BUN/creat, and urine monthly for first 3 mo and q3mo thereafter; consider baseline ophthalmology examination and radiography; should be prescribed by physicians thoroughly familiar and experienced in its use
Alcohol consumption causes acitretin (half-life 2 d) to be metabolized to etretinate (half-life 120 d and has been found in serum up to 4 y and 4 mo after discontinuation)Isotretinoin (Accutane)
Decreases sebaceous gland size and sebum production. May inhibit sebaceous gland differentiation and abnormal keratinization.
A US Food and Drug Administration–mandated registry is now in place for all individuals prescribing, dispensing, or taking isotretinoin. For more information on this registry, see iPLEDGE. This registry aims to further decrease the risk of pregnancy and other unwanted and potentially dangerous adverse effects during a course of isotretinoin therapy.Adult
40-60 mg/d PO for 4 mo
Pediatric
Not established
Toxicity may occur with vitamin A coadministration; pseudotumor cerebri or papilledema may occur; may reduce plasma levels of carbamazepine
Absolute: Pregnancy or a woman who is likely to become pregnant or who intends to become pregnant; females who cannot use reliable contraception while undergoing treatment; noncompliance with contraception; nursing mothers; concurrent use of methotrexate (increased liver toxicity) or tetracyclines (pseudotumor cerebri); documented hypersensitivity
Relative: Leukopenia; moderate-to-severe cholesterol or triglyceride elevation, significant hepatic or renal dysfunctionPregnancy
X – Contraindicated; benefit does not outweigh risk
Precautions
May decrease night vision; inflammatory bowel disease may occur; may be associated with hepatitis; exaggerated healing response of acne lesions (excessive granulation with crusting) may occur; patients with diabetes may have problems controlling their blood sugar; avoid exposure to UV light or sunlight until tolerance is achieved; discontinue if rectal bleeding, abdominal pain, or severe diarrhea occur; should be prescribed by physicians thoroughly familiar and experienced in its use
Antihistamines
These agents are used to prevent the histamine response in sensory nerve endings and blood vessels. They are more effective in preventing histamine response than in reversing it.
Diphenhydramine (Benadryl, Benylin, Diphen, AllerMax)
For symptomatic relief of pruritus and burning caused by the release of histamine.
Adult
25-50 mg PO q6-8h prn; not to exceed 400 mg/d
10-50 mg IV/IM q6-8h prn; not to exceed 400 mg/dPediatric
12.5-25 mg PO tid/qid, 5 mg/kg/d, or 150 mg/m2/d divided tid/qid; not to exceed 300 mg/d
5 mg/kg/d IV/IM or 150 mg/m2/d divided qid; not to exceed 300 mg/d