
👤 Users: 0 | 👁 Guests: 16 | 🌍 Total: 16
Congenital dyskeratosis =عسر التقرن الخلقي
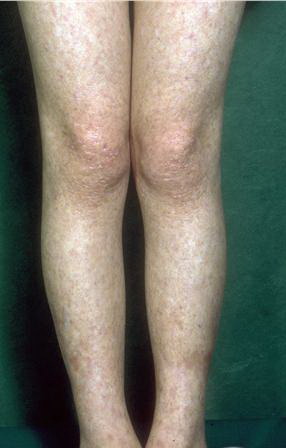
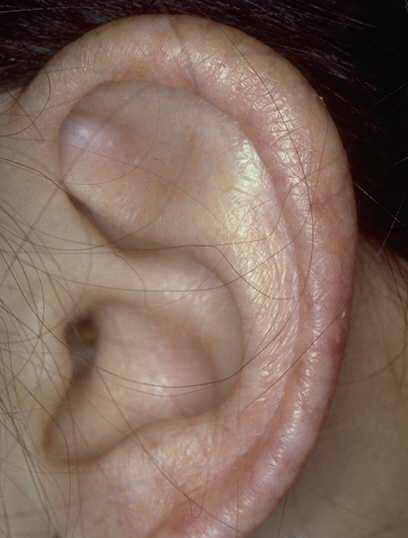
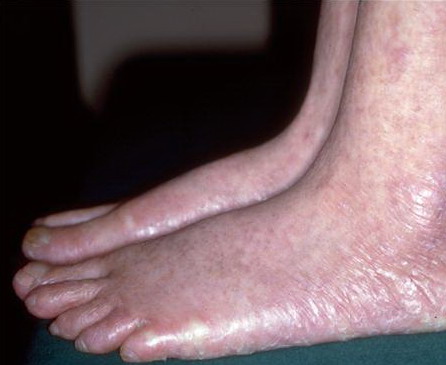
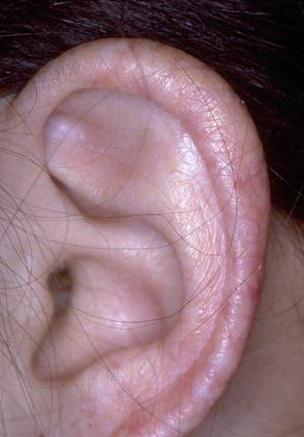
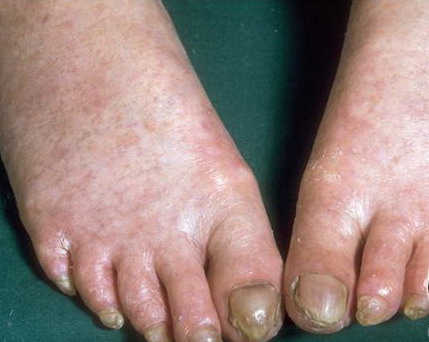
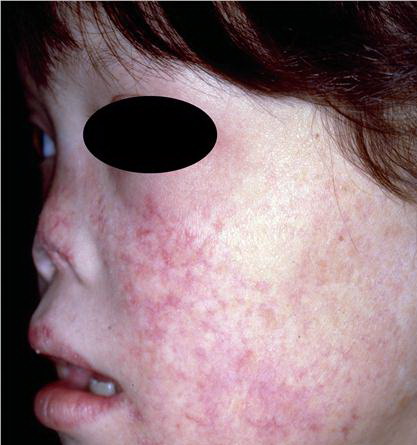
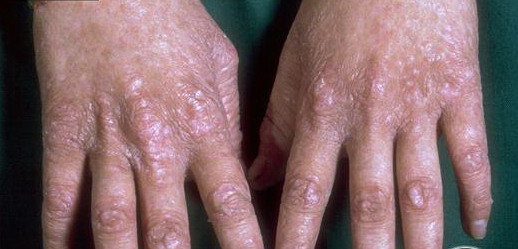
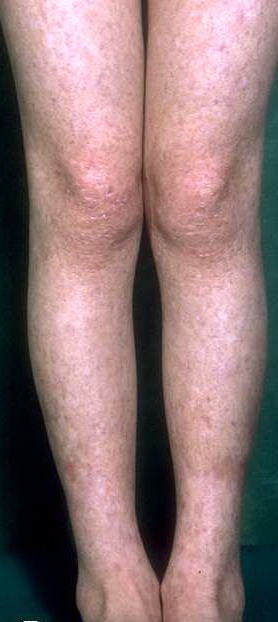
Congenital dyskeratosis
Dyskeratosis congenita (DKC), also known as Zinsser-Engman-Cole syndrome, is a rare, progressive bone marrow failure syndrome characterized by the triad of reticulated skin hyperpigmentation, nail dystrophy, and oral leukoplakia. Evidence exists for telomerase dysfunction, ribosome deficiency, and protein synthesis dysfunction in this disorder. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy.
DKC is genetically heterogeneous, with X-linked recessive (Mendelian Inheritance in Man [MIM] 305000), autosomal dominant (MIM 127550), and autosomal recessive (MIM 224230) subtypes. DKC is related to telomerase dysfunction1,2 ; all genes associated with this syndrome (ie, DKC1, TERT, TERC, NOP10) encode proteins in the telomerase complex responsible for maintaining telomeres at the ends of chromosomes. In the X-linked recessive form, the gene defect lies in the DKC1 gene (located at Xq28), which encodes for the protein dyskerin. Dyskerin is composed of 514 amino acids and has a role in ribosomal RNA processing and telomere maintenance.3,4 In the autosomal dominant form, mutations in the RNA component of telomerase (TERC) or telomerase reverse transcriptase (TERT) are responsible for disease phenotype.2,5,6
Defects in the NOP10 gene were found in association with autosomal recessive DKC.7 NOP10 encodes small nucleolar ribonucleoproteins (snoRNP) associated with the telomerase complex. In persons with autosomal dominant DKC and in terc-/- knockout mice, genetic anticipation (ie, increasing severity and/or earlier disease presentation with each successive generation) has been reported.8
Patients with DKC have reduced telomerase activity and abnormally short tracts of telomeric DNA compared with normal controls. Telomeres are repeat structures found at the ends of chromosomes that function to stabilize chromosomes. With each round of cell division, the length of telomeres is shortened and the enzyme telomerase compensates by maintaining telomere length in germline and stem cells. Because telomeres function to maintain chromosomal stability, telomerase has a critical role in preventing cellular senescence and cancer progression. Rapidly proliferating tissues with the greatest need for telomere maintenance (eg, bone marrow) are at greatest risk for failure. DKC1 has been found to be a direct target of the c-myc oncogene, strengthening the connection between DKC and malignancy.9
Analysis of 270 families in the DKC registry found that mutations in dyskerin (DKC1), TERT, and TERC only account for 64% of patients, with an additional 1% due to NOP10, suggesting that other genes associated with this syndrome are, as yet, unidentified.
The mucocutaneous features of DKC typically develop between ages 5 and 15 years. The median age of onset of the peripheral cytopenia is 10 years.
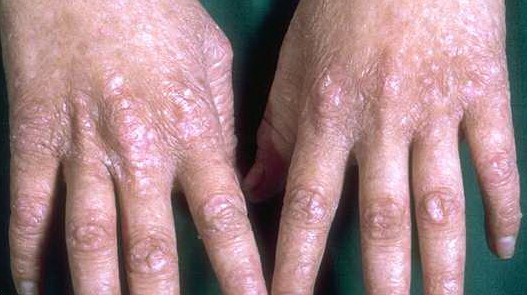
The triad of reticulated hyperpigmentation of the skin, nail dystrophy, and leukoplakia characterizes DKC. The syndrome is clinically heterogeneous; in addition to the diagnostic mucocutaneous features and bone marrow failure, affected individuals can have a variety of other clinical features.
- Cutaneous findings
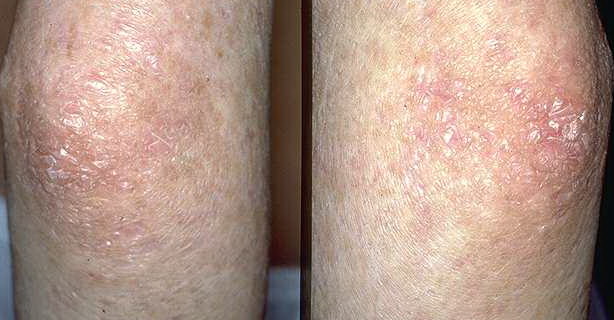
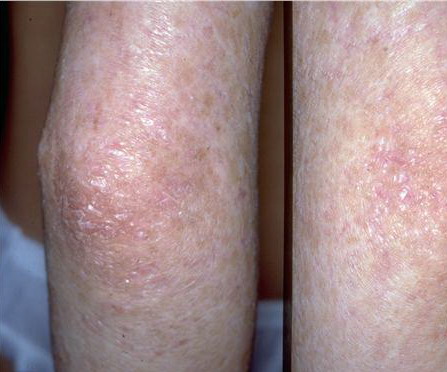
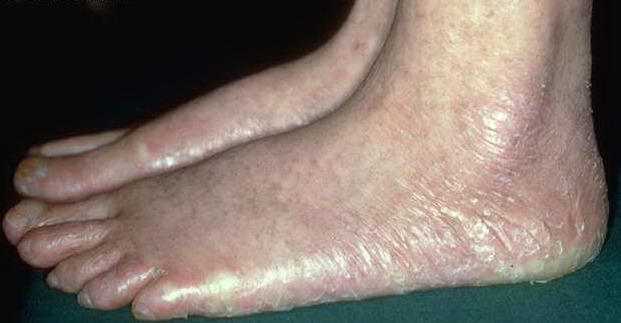
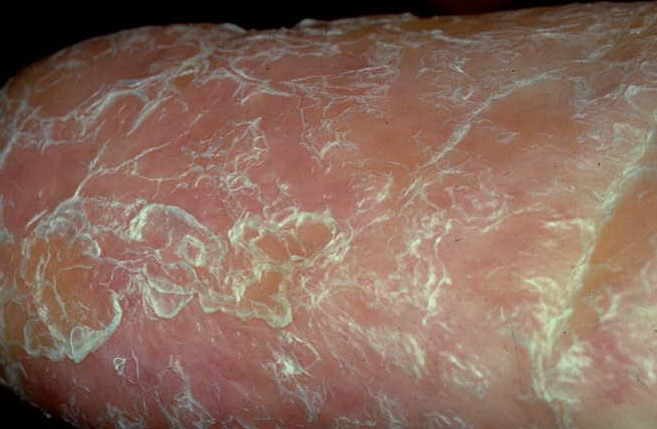
- The primary finding is abnormal skin pigmentation, with tan-to-gray hyperpigmented or hypopigmented macules and patches in a mottled or reticulated pattern. Reticulated pigmentation occurs in approximately 90% of patients. Poikilodermatous changes with atrophy and telangiectasia are common.
- The cutaneous presentation may clinically and histologically resemble graft versus host disease. The typical distribution involves the sun-exposed areas, including the upper trunk, neck, and face.
- Other cutaneous findings may include alopecia of the scalp, eyebrows, and eyelashes; premature graying of the hair; hyperhidrosis; hyperkeratosis of the palms and soles; and adermatoglyphia (loss of dermal ridges on fingers and toes).
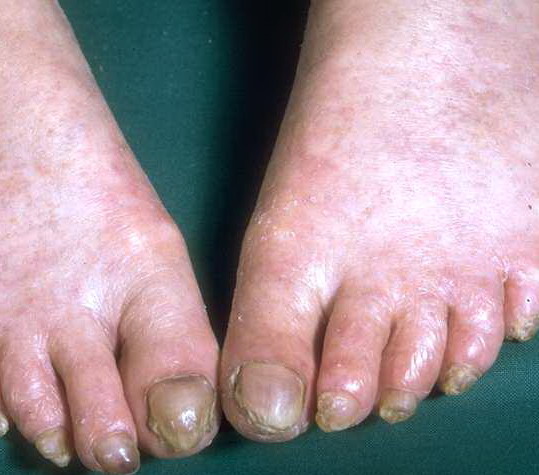
- Nail findings
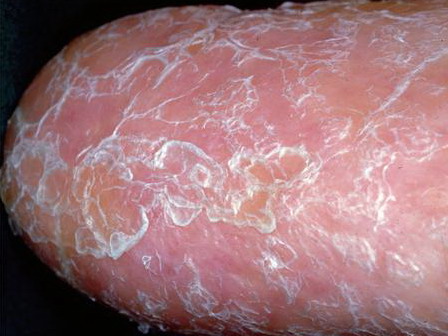
- Nail dystrophy is seen in approximately 90% of patients, with fingernail involvement often preceding toenail involvement.
- Progressive nail dystrophy begins with ridging and longitudinal splitting. Progressive atrophy, thinning, pterygium, and distortion eventuate in small, rudimentary, or absent nails.
- Mucosal findings
- Mucosal leukoplakia occurs in approximately 80% of patients and typically involves the buccal mucosa, tongue, and oropharynx. The leukoplakia may become verrucous, and ulceration may occur. Patients also may have an increased prevalence and severity of periodontal disease.
- Other mucosal sites may be involved (eg, esophagus, urethral meatus, glans penis, lacrimal duct, conjunctiva, vagina, anus). Constriction and stenosis can occur at these sites, with subsequent development of dysphagia, dysuria, phimosis, and epiphora.
- Bone marrow failure
- Approximately 90% have peripheral cytopenia of one or more lineages. In some cases, this is the initial presentation, with a median age of onset of 10 years.
- Bone marrow failure is a major cause of death, with approximately 70% of deaths related to bleeding and opportunistic infections as a result of bone marrow failure.
- Pulmonary complications
- Approximately 20% of individuals with DKC develop pulmonary complications, including pulmonary fibrosis and abnormalities of pulmonary vasculature.
- The recommendation is that DKC patients avoid taking drugs with pulmonary toxicity (eg, busulfan) and that they have their lungs shielded from radiation during BMT.
- Increased risk of malignancy
- Patients have an increased prevalence of malignant mucosal neoplasms, particularly squamous cell carcinoma of the mouth, nasopharynx, esophagus, rectum, vagina, or cervix. These often occur within sites of leukoplakia.
- The prevalence of squamous cell carcinoma of the skin is also increased. Other malignancies reported include Hodgkin lymphoma, adenocarcinoma of the gastrointestinal tract, and bronchial and laryngeal carcinoma.
- Malignancy tends to develop in the third decade of life.
- Neurologic system findings: Patients may have learning difficulties and mental retardation.
- Ophthalmic system findings: DKC reportedly is associated with conjunctivitis, blepharitides, and pterygium. Lacrimal duct stenosis resulting in epiphora (ie, excessive tearing) occurs in approximately 80% of patients.
- Skeletal system findings: Patients may have mandibular hypoplasia, osteoporosis, avascular necrosis, and scoliosis.
- Gastrointestinal system findings: These may include esophageal webs, hepatosplenomegaly, and cirrhosis.
- Genitourinary system findings: Hypospastic testes, hypospadias, and ureteral stenosis are reported.
- Female carriers: Female carries of DKC may have subtle clinical features. One study showed that 3 of 20 female carriers had clinical features that included a single dystrophic nail, a patch of hypopigmentation, or mild leukoplakia.
Mutations in DKC1 have been shown to cause the X-linked form of DKC. The inheritance pattern of most cases of DKC is X-linked recessive, but autosomal dominant and recessive patterns have been reported. Autosomal dominant DKC is associated with TERC and TERT mutations in some cases, and NOP10 has been associated with some cases of autosomal recessive DKC.
Laboratory Studies
Perform appropriate tests to screen for bone marrow failure, pulmonary disease, neurologic disease, and mucosal malignancies. Specific tests depend on the clinical findings and may include a CBC count, chest radiography, pulmonary function tests, and stool tests for occult blood. Elevated von Willebrand factor levels have been associated with fatal vascular complications after BMT and may be a marker for patients with a predisposition for endothelial deterioration.
Mutational analysis may be useful in confirming the diagnosis. Mutations in the TERC gene and in the TERT gene, the gene for telomerase reverse transcriptase (another member of the ribonucleoprotein complex), have been identified in a subset of patients with aplastic anemia.10 Genetic testing for occult DKC should be considered in patients with aplastic anemia. However, a 2006 genetic analysis of the TERC gene among 284 children with either aplastic anemia or myelodysplastic syndrome found only 2 mutations in the TERC gene.11
Patients and family members without a known mutation can be screened with a new test, leukocyte subset flow fluorescence in situ hybridization, which can identify very short telomeres in both clinically apparent and silent disease.12
Several reports note that radiographs show calcification of the basal ganglia.
Histologic Findings
Skin biopsy specimens from the areas of reticulated pigmentation typically show nonspecific changes, including mild hyperkeratosis, epidermal atrophy, telangiectasia of the superficial blood vessels, and melanophages in the papillary dermis. Interface changes have also been reported, with mild basal layer vacuolization and a lymphocytic inflammatory infiltrate in the upper dermis.
Medical Care
Short-term treatment options for bone marrow failure in patients with DKC include anabolic steroids (eg, oxymetholone), granulocyte macrophage colony-stimulating factor, granulocyte colony-stimulating factor, and erythropoietin13 ; however, the only long-term, curative option is hematopoietic stem cell transplantation (SCT).
- Approximately 50% of patients experience a temporary increase in blood counts with androgen therapy; the duration of treatment is limited by adverse effects; in addition, reports have described splenic peliosis and rupture in patients treated concomitantly with androgens and granulocyte colony-stimulating factor.14
- The success rate of SCT is limited because of a high prevalence of fatal pulmonary complications, which likely reflect preexisting pulmonary disease in these patients.
- Drugs that cause pulmonary toxicity (eg, busulfan) and exposure to unnecessary radiation should be avoided in these patients.
- Nonmyeloablative hematopoietic SCT conditioning regimens (ie, reduced-intensity conditioning) with fludarabine may offer better outcomes. A 2007 review showed a 22% mortality rate with reduced-intensity conditioning in DKC treatment versus a 71% mortality rate with traditional myeloablative regimens.15
- The best candidates for transplantation may be patients with sibling donors and with no preexisting pulmonary disease.
The elucidation of the genetic basis of X-Iinked DKC enables prenatal testing and carrier detection. Early diagnosis of DKC through genetic analysis also may help identify patients for early harvest and storage of their bone marrow for use after anticipated marrow failure. In the future, patients with DKC may be candidates for hematopoietic gene therapy.
Medication
The goals of pharmacotherapy are to reduce morbidity and to prevent complications.
Colony-stimulating factors
Used to stimulate bone marrow in patients with cytopenia of one or more cell lineage.
Erythropoietin (Epogen, Procrit)
Stimulates division and differentiation of erythroid progenitor cells.
- Dosing
- Interactions
- Contraindications
- Precautions
Adult
50-100 U/kg IV/SC, 3 times/wk; dosing may vary
Pediatric
Not established
- Dosing
- Interactions
- Contraindications
- Precautions
None reported
- Dosing
- Interactions
- Contraindications
- Precautions
Documented hypersensitivity; uncontrolled hypertension
- Dosing
- Interactions
- Contraindications
- Precautions
Pregnancy
C – Fetal risk revealed in studies in animals but not established or not studied in humans; may use if benefits outweigh risk to fetus
Precautions
Caution in porphyria, hypertension, and history of seizures; decrease dose if hematocrit value increase exceeds 4 U in any 2-wk period
Filgrastim (Neupogen)
Activates and stimulates production, maturation, migration, and cytotoxicity of neutrophils.
- Dosing
- Interactions
- Contraindications
- Precautions
Adult
5 mcg/kg/d SC; dosing may vary
Pediatric
Not established