
👤 Users: 0 | 👁 Guests: 1 | 🌍 Total: 1
Amyloidosis-Nodular= الداء النشواني العقيدي
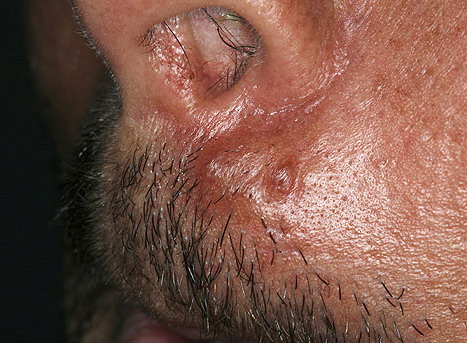
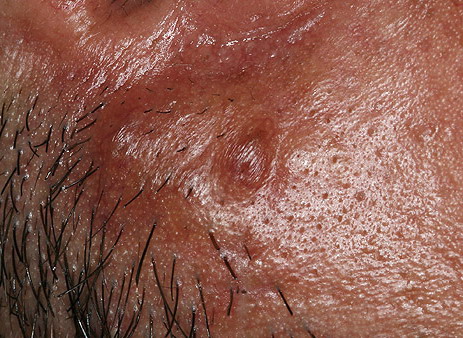
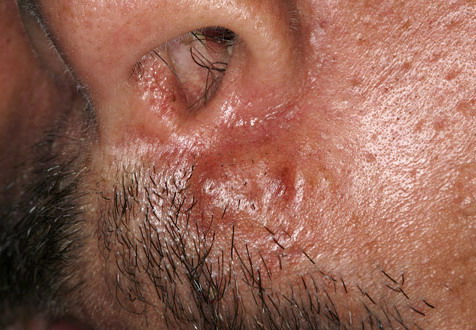
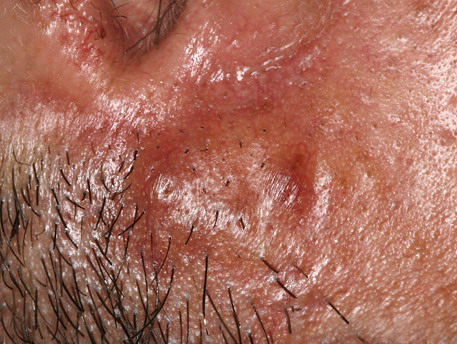
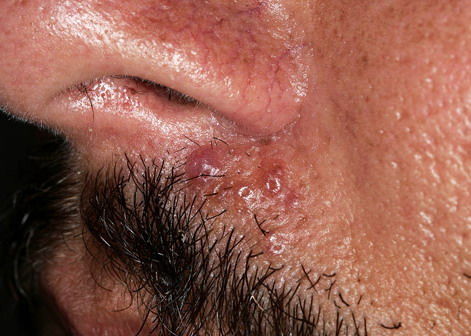
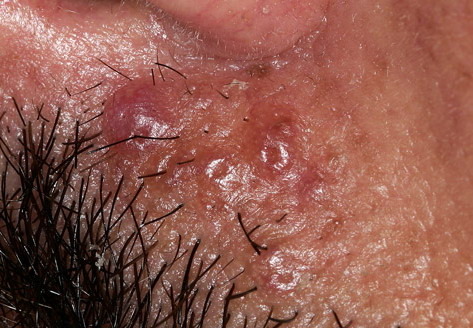
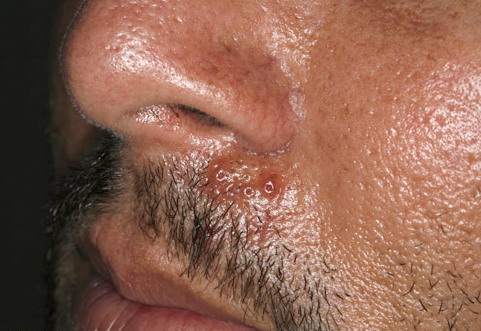
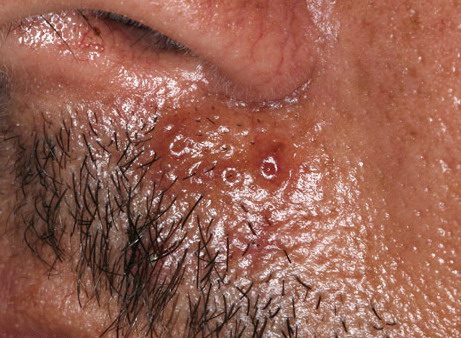
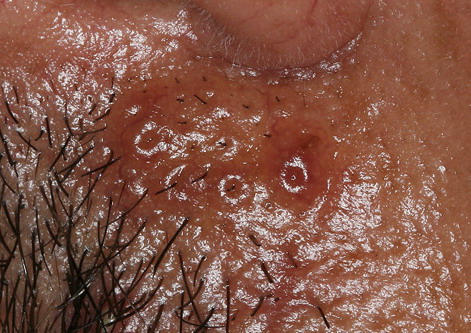
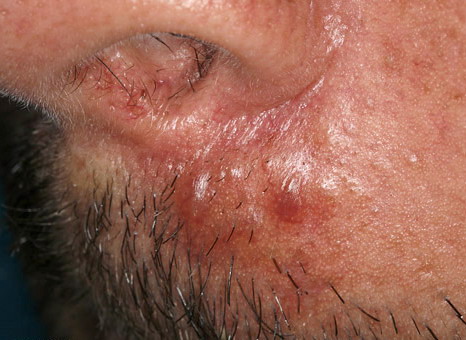
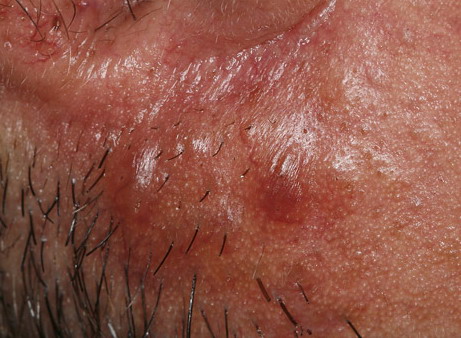
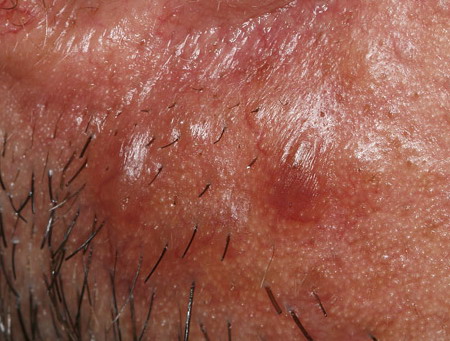
Nodular Amyloidosis
Localized cutaneous amyloidosis (LCA) refers to a condition characterized by the deposition of amyloid or amyloid-like proteins in the dermis. Localized cutaneous amyloidosis encompasses several conditions characterized by amyloid deposition, including macular amyloidosis and lichen amyloidosis. Nodular localized cutaneous amyloidosis (NLCA) is the rarest type of localized cutaneous amyloidosis and is distinct from the other two.
Gottron first reported nodular localized cutaneous amyloidosis in 1950. Since then, approximately 60 patients have been reported in the North American, European, and Asian literature. This entity also is termed amyloidosis cutis nodularis atrophicans or tumefactive amyloid. By definition, nodular localized cutaneous amyloidosis describes a primary disease of the skin, although lesions occasionally appear similar to the skin manifestations of systemic amyloidosis.
Pathophysiology
As a term, “amyloid” was used historically to define proteins that shared similar microscopic characteristics and affinity for certain stains. Research has revealed that “amyloid” proteins are heterogeneous. The various diseases characterized by deposition of “amyloid” proteins are similarly heterogeneous but have in common the deposits of fibrillar proteins characterized as “amyloid” in the dermis. In nodular localized cutaneous amyloidosis, the amyloid is believed to derive from local plasma cells, in contrast to lichenoid or macular amyloidosis, which have keratinocyte-derived amyloid.
In nodular localized cutaneous amyloidosis, plasma cells produce immunoglobulin light chains that are precursors to the amyloid fibril protein(s) termed amyloid L. Reports differ regarding the clonality of this population of plasma cells. In some instances, plasma cells have been monoclonal, suggesting that nodular localized cutaneous amyloidosis is a neoplastic disorder1 ; however, in another instance, plasma cells demonstrated polyclonality, which usually is a feature of a more reactive process.
Nodular localized cutaneous amyloidosis typically is benign and limited to the skin. However, lesions are more often persistent. Reported rates of progression to systemic disease are derived from case series with small numbers of patients; these rates vary from 7% to nearly 50%.2,3 As many as 25% of reported cases have been associated with Sjögren syndrome. Some speculate that these 2 disorders have may have a shared pathogenesis
History
- Nodular localized cutaneous amyloidosis lesions usually are asymptomatic.
- Patients can present with single or multiple lesions.
- In some reports, lesions were present for several years before patients sought medical attention.
- Troublesome aspects of nodular localized cutaneous amyloidosis primarily result from patient concerns about appearance, although plaques eventually fissured in one patient in whom the plantar aspects of the feet were affected.
- Up to 25% of reported cases of NLCA have been in patients with Sjögren syndrome.
Physical
- Firm nodules can present anywhere on the skin, including the face, scalp, extremities, trunk, and genitalia.10,11,12
- Nodules vary from a few millimeters to a few centimeters.
- Nodules appear pink to brown or red.
- Overlying epidermal atrophy has been described.
- Other terms that describe the various lesions of nodular localized cutaneous amyloidosis include waxy, purpuric, yellowish, or bullous.
- Lesions tend not to ulcerate.
- Nodular localized cutaneous amyloidosis lacks extracutaneous findings by definition; however, one patient reported to have nodular localized cutaneous amyloidosis had amyloid deposits in the rectum.
- Macroglossia, a typical feature of systemic amyloidosis, is not seen in nodular localized cutaneous amyloidosis.
Causes
The cause of nodular localized cutaneous amyloidosis is not known, although the amyloid protein is derived from a localized infiltrate of plasma cells.
Laboratory Studies
- Normal serum protein electrophoresis and urine protein electrophoresis studies help to exclude multiple myeloma, which can also cause amyloid deposits made up of immunoglobulin light chains.
- Positive antinuclear, anti-Ro, and anti-La antibodies suggest Sjögren syndrome.
- Laboratory studies, such as CBC, serum chemistry profile, and liver function tests often were part of a general workup in several case reports of patients with nodular localized cutaneous amyloidosis. Nodular localized cutaneous amyloidosis does not cause any abnormal findings in these studies.
- Urinalysis or 24-hour urine testing can be performed to check for protein. Proteinuria is not a feature of localized cutaneous disease but can be seen in systemic amyloidosis.
Imaging Studies
- In some patients, imaging studies have included chest radiography, ECG, and abdominal ultrasonography.
- Screening for amyloid within organs can be accomplished using scintigraphy with radioiodinated serum amyloid P component (ie, SAP scanning). This is a very sensitive test for detecting early systemic amyloidosis.
Procedures
- Skin biopsy provides the definitive diagnosis. No special tissue preparation or handling is required before delivering the specimen to the laboratory. Special stains and immunohistochemistry are helpful.
- An optimal biopsy specimen includes the epidermis, papillary dermis, and reticular dermis. The amyloid in nodular localized cutaneous amyloidosis is located in the reticular dermis and subcutaneous fat, and clearly differentiates nodular localized cutaneous amyloidosis from other forms of amyloidosis. A shave biopsy or other superficial sample may not include enough reticular dermis to complete the diagnosis.
- Consider bone marrow biopsy with gene rearrangement studies (if available) to exclude multiple myeloma.
Histologic Findings
Despite their biochemical heterogeneity, all “amyloid” deposits demonstrate a similar light microscopic appearance. They are eosinophilic and homogeneous when stained with hematoxylin and eosin and viewed with standard optics. When stained with Congo red and viewed with polarized light, deposits exhibit a characteristic green birefringence. In nodular amyloidosis, amyloid is not limited to the papillary dermis but is present in the entire dermis and may extend to subcutaneous fat. Amyloid deposition may be particularly prominent in walls of small blood vessels and surrounding individual lipocytes (see the images)
Plasma cells, which most likely produce the amyloid, occur within an adjacent and intermingled inflammatory infiltrate. They can be sparse or numerous (similar plasma cell infiltrate occurs in nodular pulmonary amyloidosis but usually is absent in cutaneous lesions of primary systemic amyloidosis). When eosinophilic amyloid material is exposed to potassium permanganate prior to staining with Congo red, the amyloid retains its congophilia, similar to systemic amyloidosis but in contradistinction to secondary amyloidosis. Kappa or lambda light chains (or both) may be present on immunohistochemical staining.13
When viewed with a transmission electron microscope, the apparently homogeneous deposits of amyloid are composed of loosely interwoven 6- to 10-nm–thick straight filaments. The amino acids of the filament proteins are arranged in a characteristic beta-pleated sheet tertiary structure. Amyloid deposits in the skin also contain small amounts of a plasma-derived, nonfibrillar, amyloid-P protein.
Plasma cells, which most likely produce the amyloid, occur within an adjacent and intermingled inflammatory infiltrate. They can be sparse or numerous (similar plasma cell infiltrate occurs in nodular pulmonary amyloidosis but usually is absent in cutaneous lesions of primary systemic amyloidosis). When eosinophilic amyloid material is exposed to potassium permanganate prior to staining with Congo red, the amyloid retains its congophilia, similar to systemic amyloidosis but in contradistinction to secondary amyloidosis. Kappa or lambda light chains (or both) may be present on immunohistochemical staining.13
When viewed with a transmission electron microscope, the apparently homogeneous deposits of amyloid are composed of loosely interwoven 6- to 10-nm–thick straight filaments. The amino acids of the filament proteins are arranged in a characteristic beta-pleated sheet tertiary structure. Amyloid deposits in the skin also contain small amounts of a plasma-derived, nonfibrillar, amyloid-P protein.
Medical Care
Various methods attempt to improve the appearance of the nodular localized cutaneous amyloidosis lesions, including topical and intralesional corticosteroids, cryotherapy, dermabrasion,14 shaving, curettage and electrodesiccation, carbon dioxide laser,15,16 and pulsed dye laser.17 However, lesions frequently recur after treatment. Topical and intralesional corticosteroids and cryotherapy usually are not helpful. One attempt at cryotherapy produced pinpoint bleeding.
Surgical Care
- Procedures such as excision and curettage and electrodesiccation have provided satisfactory cosmetic results for nodular localized cutaneous amyloidosis.18
- Laser treatment has been described in nodular localized cutaneous amyloidosis.
- Excessive tissue friability and difficulty with intraoperative hemostasis were described while treating one nasal lesion with carbon dioxide laser; however, a good cosmetic result was achieved.
- A patient treated with a tunable dye laser had a good result, and clinical improvement was maintained over 6 months.
- None of these treatment methods totally eradicates lesions, which can recur.
Prognosis
- Nodular localized cutaneous amyloidosis typically is benign and limited to the skin. Progression to systemic disease is unlikely. Previous articles have reported progression rates as high as 50%.2 However, a more recent review article reports that less than 15% of patients later developed systemic amyloidosis. Other, more recent case series have shown rates of progression of 7%