
Contact Us :
👤 Users: 0 | 👁 Guests: 2 | 🌍 Total: 2
Autoimmune thrombocytopenic purpura = الفرفرية بنقص الصفيحات المناعي الذاتي
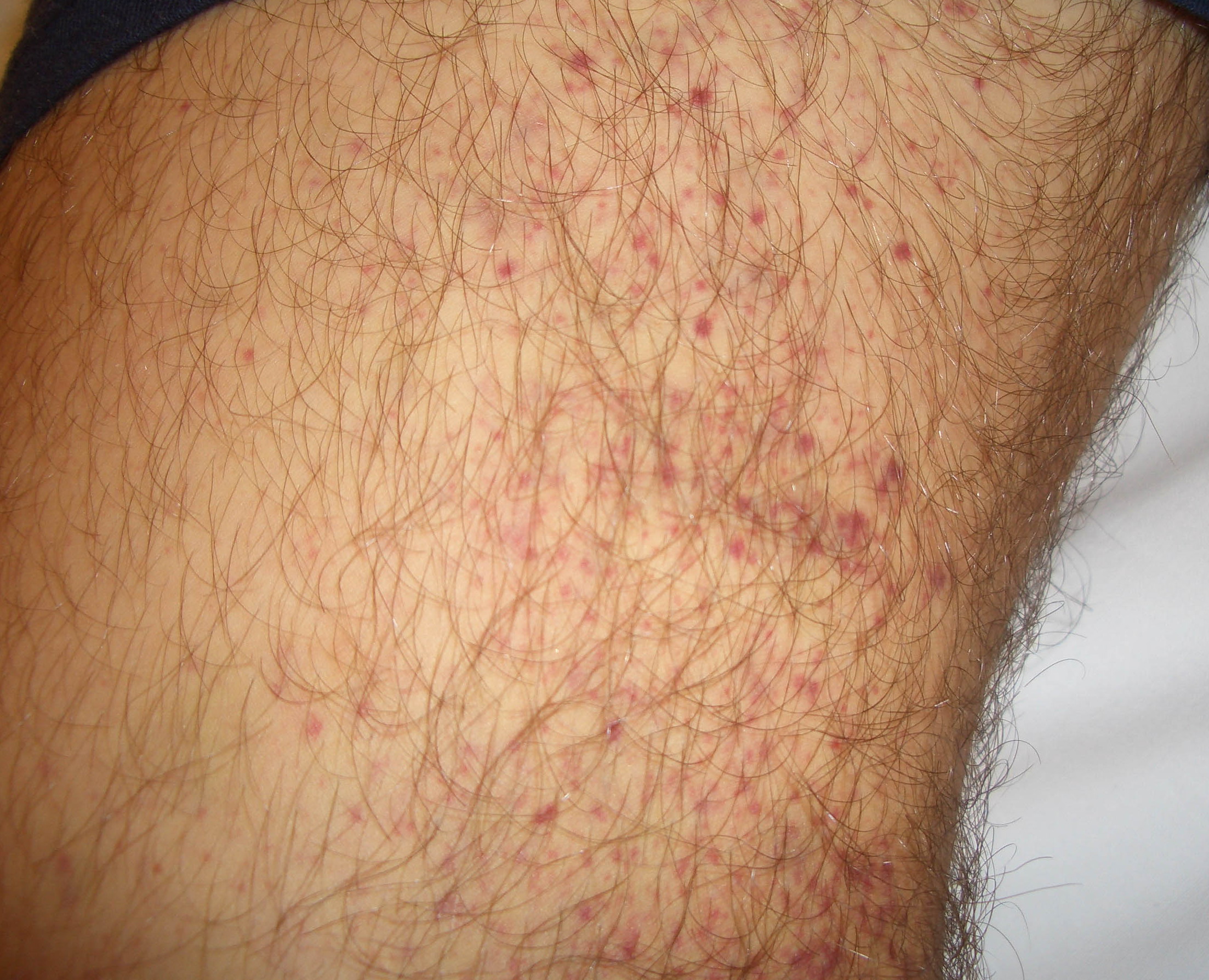
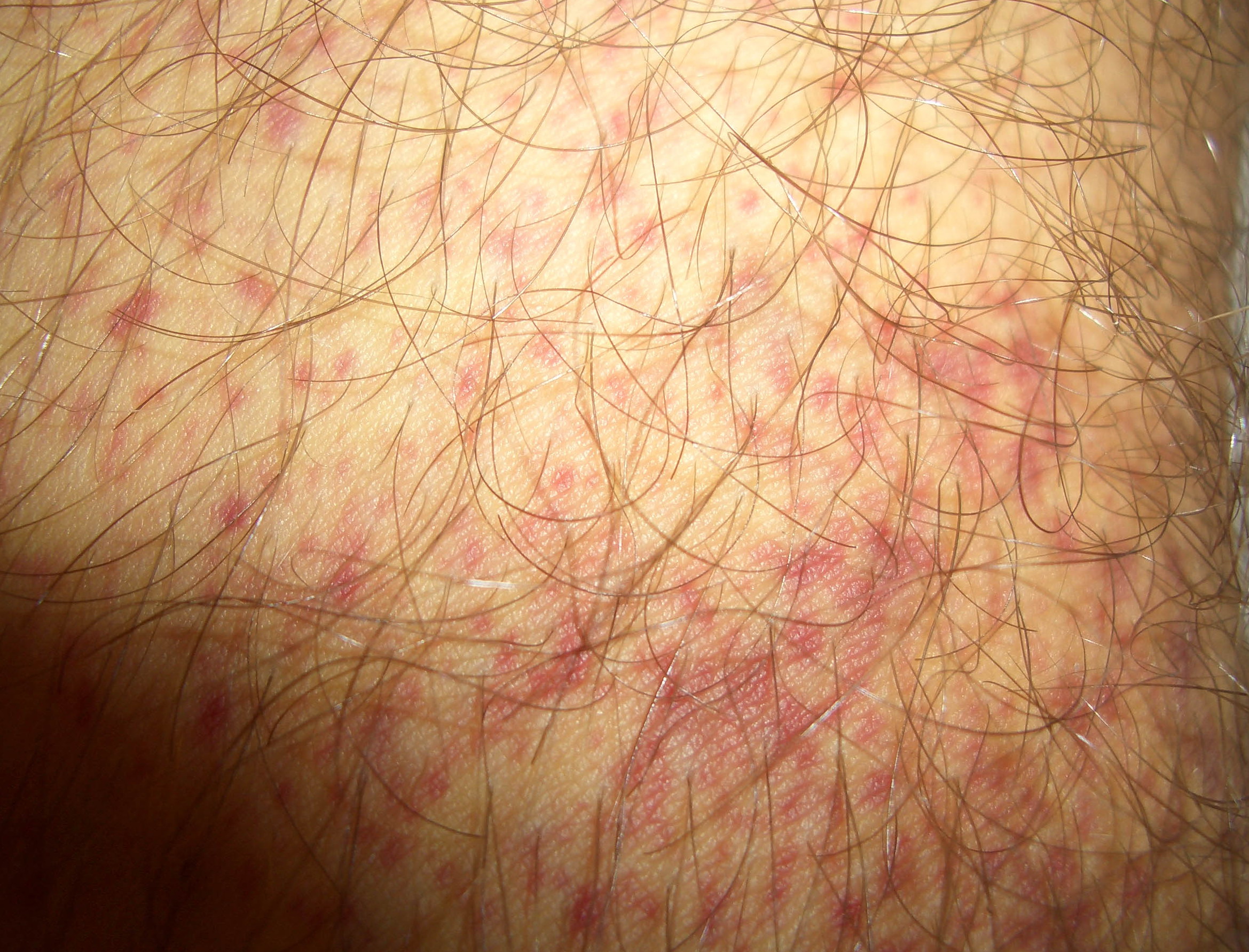
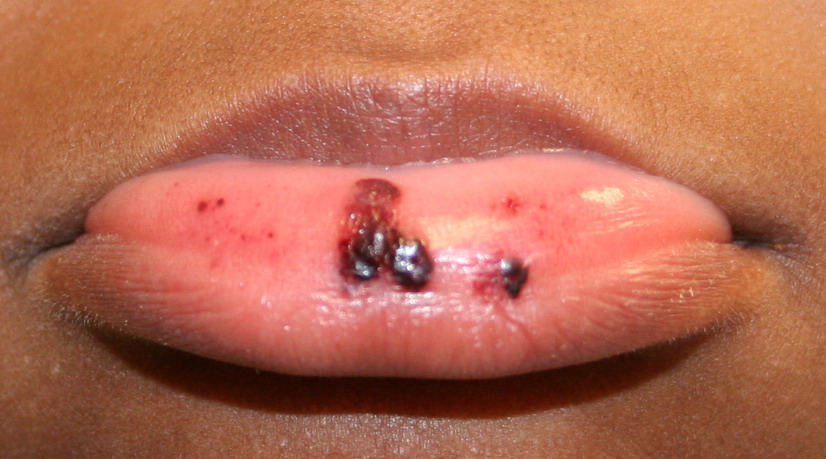
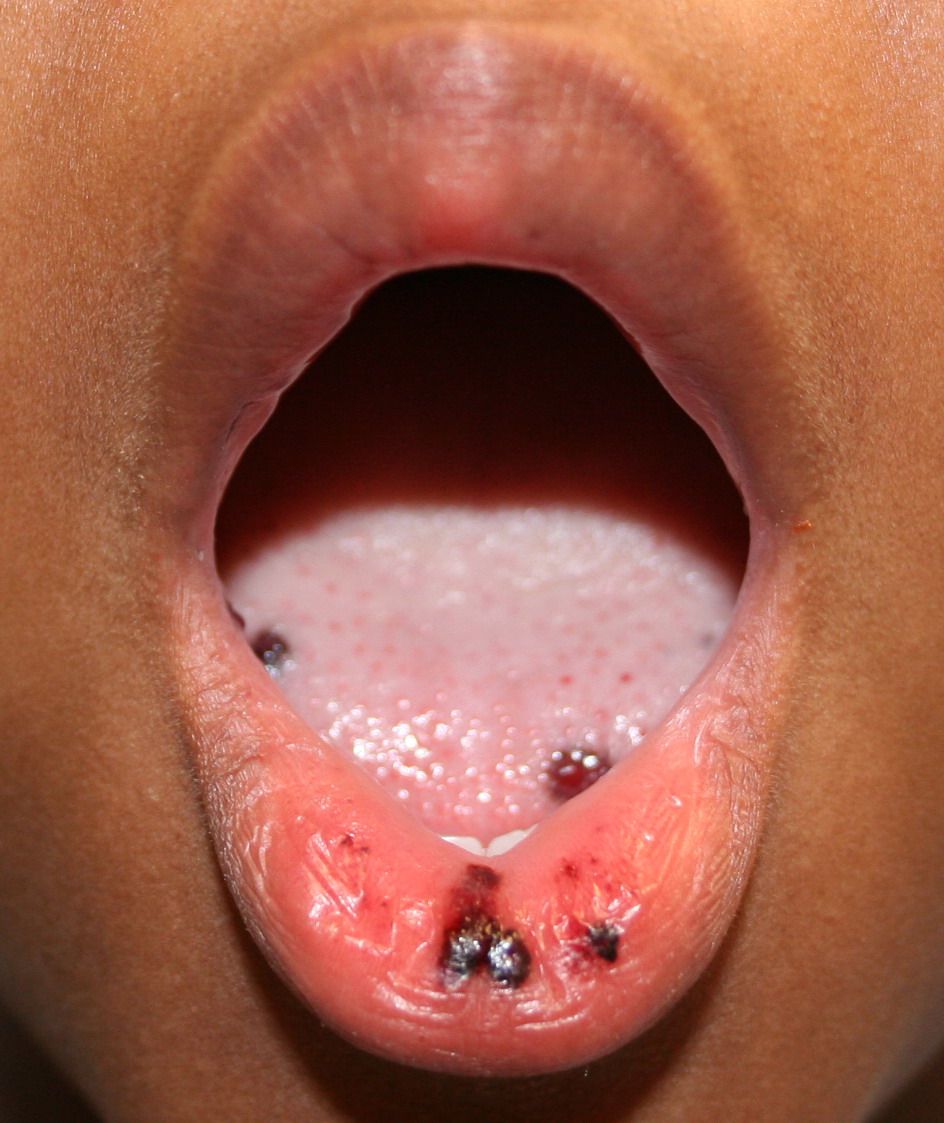
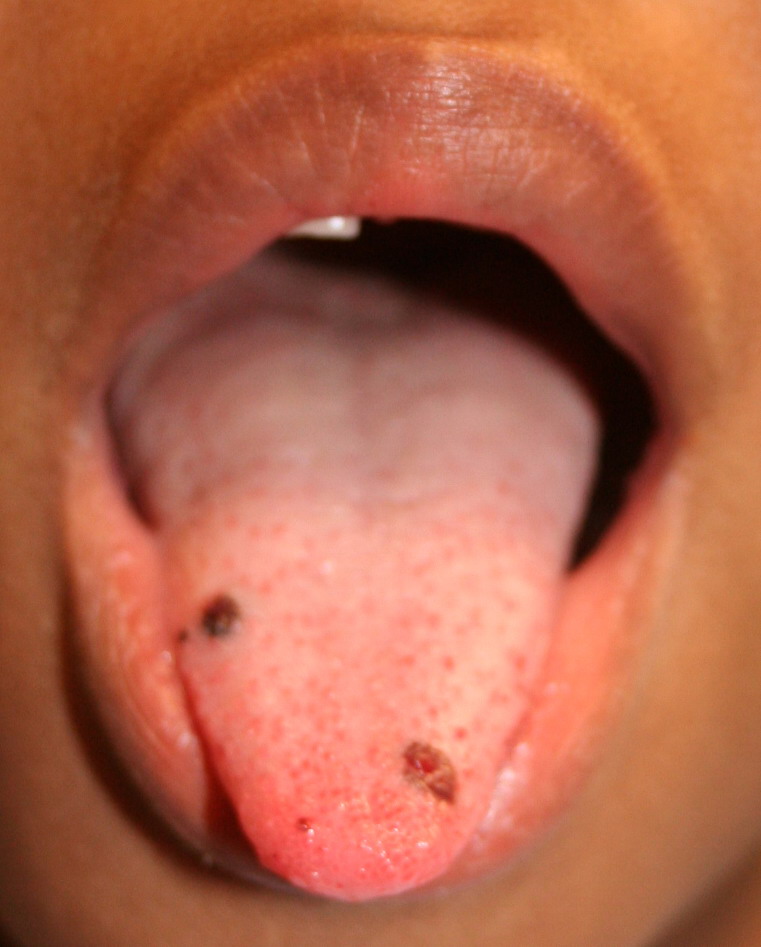
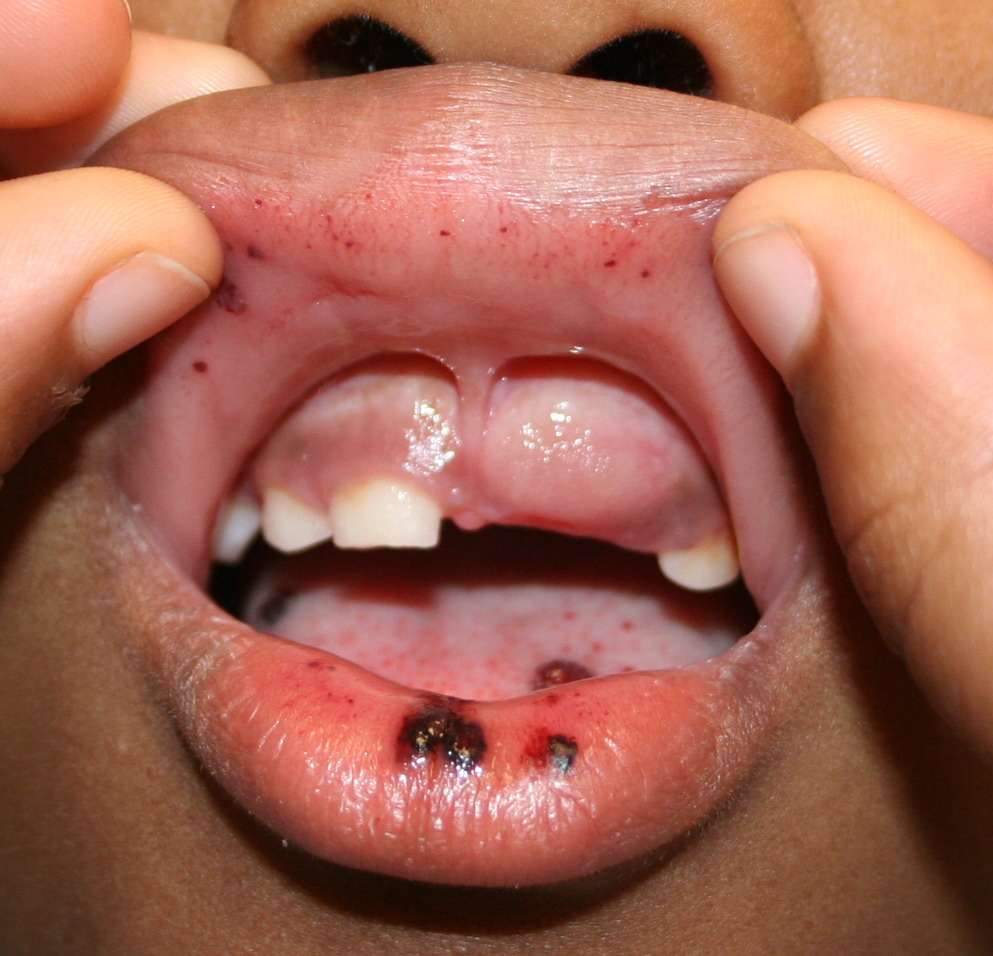
Autoimmune thrombocytopenic purpura
Idiopathic thrombocytopenic purpura (ITP), also known as primary immune thrombocytopenic purpura and autoimmune thrombocytopenic purpura, is defined as isolated thrombocytopenia with normal bone marrow and the absence of other causes of thrombocytopenia. The 2 distinct clinical syndromes manifest as an acute condition in children and a chronic condition in adults.
ITP is a decrease in the number of circulating platelets in the absence of toxic exposure or a disease associated with a low platelet count.
Pathophysiology
ITP is primarily a disease of increased peripheral platelet destruction, with most patients having antibodies to specific platelet membrane glycoproteins. Relative marrow failure may contribute to this condition, since studies show that most patients have either normal or diminished platelet production.
Acute ITP often follows an acute infection and has a spontaneous resolution within 2 months. Chronic ITP persists longer than 6 months without a specific cause.
Frequency
The incidence of ITP in adults is approximately 66 cases per 1,000,000 per year.
An average estimate of the incidence in children is 50 cases per 1,000,000 per year.
New cases of chronic refractory ITP comprise approximately 10 cases per 1,000,000 per year.
According to studies in Denmark and England, childhood ITP occurs in approximately 10-40 cases per 1,000,000 per year. A study in Kuwait reported a higher incidence of 125 cases per 1,000,000 per year.
- Hemorrhage represents the most serious complication; intracranial hemorrhage is the most significant. The mortality rate from hemorrhage is approximately 1% in children and 5% in adults. In patients with severe thrombocytopenia, predicted 5-year mortality rates from bleeding are significantly raised in patients older than 60 years versus patients younger than 40 years, 47.8% versus 2.2%, respectively.
- Older age and previous history of hemorrhage increase the risk of severe bleeding in adult ITP.
- Spontaneous remission occurs in more than 80% of cases in children but is uncommon in adults.
- In chronic ITP (adults), the female-to-male ratio is 2.6:1. More than 72% of patients older than 10 years are female.
- In acute ITP (children), distribution is equal between males (52%) and females (48%).
- Peak prevalence occurs in adults aged 20-50 years.
- Peak prevalence occurs in children aged 2-4 years.
- Approximately 40% of all patients are younger than 10 years.
- Focus on the symptoms of bleeding (eg, type, severity, duration) and on symptoms that may exclude other causes of thrombocytopenia.
- Elicit risk factors for HIV and systemic symptoms linked to other illnesses or to medications (eg, heparin, alcohol, quinidine/quinine, sulfonamides) that may cause thrombocytopenia.
- Address risk factors for increased bleeding, such as GI disease, CNS disease, urologic disease, or active lifestyle, as these may determine the aggressiveness of management.
- Common signs, symptoms, and precipitating factors include the following:
- Abrupt onset (childhood ITP)
- Gradual onset (adult ITP)
- Purpura
- Menorrhagia
- Epistaxis
- Gingival bleeding
- Recent live virus immunization (childhood ITP)
- Recent viral illness (childhood ITP)
- Bruising tendency
- Limited data are available on the recurrent form of the disease. One study showed a 6% prevalence of recurrent ITP with most patients (69%) having only one recurrence. Though one third of patients had their recurrent episode within 3 months of their initial one, the remainder of patients had at least a 3-month interval between episodes.
Physical
Evaluate the type and the severity of bleeding and try to exclude other causes of bleeding. Seek evidence of liver disease, thrombosis, autoimmune diseases (eg, nephritis, cutaneous vasculitis, arthritis), and infection, particularly HIV.
Common physical findings include the following:
- Nonpalpable petechiae, which mostly occur in dependent regions
- Hemorrhagic bullae on mucous membranes
- Purpura
- Gingival bleeding
- Signs of GI bleeding
- Menometrorrhagia, menorrhagia
- Retinal hemorrhages
- Evidence of intracranial hemorrhage, with possible neurologic symptoms
- Nonpalpable spleen: The prevalence of palpable spleen in patients with ITP is approximately the same as that in the non-ITP population (ie, 3% in adults, 12% in children).
- Spontaneous bleeding when platelet count is less than 20,000/mm3.
Causes
- Immunoglobulin G (IgG) autoantibodies on the platelet surface
Treatment
Prehospital Care
- Prehospital care focuses on the ABCs, which include providing oxygen, controlling severe hemorrhage, and initiating intravenous (IV) fluids to maintain hemodynamic stability.
- Prehospital airway control may be necessary for a large intracranial hemorrhage.
- EMS providers should be aware of the potential for serious bleeding complications in patients with idiopathic thrombocytopenic purpura (ITP).
Emergency Department Care
- Life-threatening bleeding requires conventional critical care interventions.
- In the patient with known ITP, high-dose parenteral glucocorticoids and IV immunoglobulin (IVIg), with or without platelet transfusions, are appropriate.
- Platelet transfusion is indicated for controlling severe hemorrhage. Send a blood specimen to the lab for type and screen in case platelet transfusion is necessary.
- Platelet survival is increased if the platelets are transfused immediately after IVIg infusion.
- A consultation with a hematologist may be required to make a decision regarding the transfusion of platelets.
- Guidelines for transfusion dosage
- 6-8 U of platelet concentrate, or 1 U/10 kg
- 1 U of platelets to increase count of a 70-kg adult by 5-10,000/mm3 and an 18-kg child by 20,000/mm3
- Splenectomy is reserved for patients in whom medical therapy fails. Emergent splenectomy is indicated in patients with life-threatening bleeding in whom medical therapy fails.
- In patients without life-threatening complications, focus ED care on confirming the diagnosis, if possible, and initiating therapy as needed.
- Most patients with undiagnosed thrombocytopenia and purpura will need admission for further evaluation and treatment, since ITP is a diagnosis of exclusion.
Consultations
- Consult a hematologist for assistance in confirming the diagnosis or, in the patient with known ITP, arranging disposition and follow-up care, if appropriate.
- Consult a neurosurgeon for intracranial hemorrhage. Consultation by other surgical specialists may be required for extensive hemorrhage at other sites
Medication
- .
Glucocorticoids and IVIg are the mainstays of medical therapy. Indications for use, dosage, and route of administration are based on the patient’s clinical condition, the absolute platelet count, and the degree of symptoms. Consultation with a hematologist may be needed prior to starting therapy.
Children who have platelet counts >30,000/mm3 and are asymptomatic or have only minor purpura do not require routine treatment. Children who have platelet counts <20,000/mm3 and significant mucous membrane bleeding and those who have platelet counts <10,000/mm3 and minor purpura should receive specific treatment.
Adults with platelet counts >50,000/mm3 do not require treatment. Treatment is indicated for adults with counts <50,000/mm3 with significant mucous membrane bleeding. Treatment also is indicated for those adults with risk factors for bleeding (eg, hypertension, peptic ulcer disease, vigorous lifestyle) and in patients with a platelet count <20,000-30,000/mm3.
IV anti-(Rh)D, also known as IV Rh immune globulin (IG), was not recommended by the 1996 American Society of Hematology practice guidelines. However, recent studies using higher dosages of IV RhIG in acute ITP in children and adults show platelet count increases at 24 hours faster than medicating with steroids and at 72 hours similar to IVIg. Although generally less toxic than IV steroids, IV RhIG is more expensive than IV steroids. Studies in children with chronic ITP show that escalating or elevated doses of IV RhIG have comparable responses to those of high-dose IVIg therapy in children. This therapy is not appropriate for patients who have undergone splenectomy. Acute intravascular hemolysis after infusing IV RhIG has been reported, with an estimated incidence of 1 in 1115 patients.
Steroid use and immunosuppressives and splenectomy may be undesirable because of their associated complications. For long-term steroid use, this includes osteoporosis, glaucoma, cataracts, loss of muscle mass, and an increased risk of infection. For immunosuppressive therapy and splenectomy, risks include worsening immunosuppression and infection or sepsis. Studies of the use of multiagent therapies in refractory patients are ongoing. Some small studies have shown limited success. According to one study1 , using a combination of weekly vincristine, weekly methylprednisolone, both until platelet counts reached 50,000/mm3, and cyclosporine orally twice daily until the platelet count is normal for 3-6 months seems promising, though larger prospective studies are needed.
Other therapies, such as cyclophosphamide, danazol, dapsone, interferon alfa, azathioprine, vinca alkaloids, accessory splenectomy, and splenic radiation have been studied. Many case series discussing these treatments are too small to show sufficient evidence of a clinically significant reduction in bleeding or mortality rate; however, they serve as additional therapeutic measures in ITP refractory-to-primary therapy (eg, glucocorticoids, IVIg immunoglobulin, splenectomy). Newer studies on rituximab suggest that this agent is an effective treatment option in splenectomized refractory or relapsed ITP patients.2,3
Clinical trials have shown promise for agents that directly stimulate platelet production, such as thrombopoietin (TPO) receptor-binding agents. Two new agents, eltrombopag and romiplostim, are available to patients with chronic ITP who have failed other therapies.4,5 Both of these agents require registration in a database. While they show promise for raising platelet counts, there are potential safety concerns such as thrombocytosis and rebound thrombocytopenia. It is unlikely that emergency physicians should be prescribing these agents without being under the recommendation of a hematologist.
Glucocorticoids
These agents are used to treat idiopathic and acquired autoimmune disorders. They have been shown to increase platelet count in ITP.
Prednisone (Deltasone, Orasone, Sterapred)
Useful in treating inflammatory and allergic reactions; may decrease inflammation by reversing increased capillary permeability and suppressing PMN activity. DOC for all adult patients with platelet counts <50,000/mm3. Asymptomatic patients with platelet counts >20,000/mm3, or patients with counts 30,000-50,000/mm3 with only minor purpura, may not need therapy; withholding medical therapy may be appropriate for asymptomatic patients, regardless of count.
- Dosing
- Interactions
- Contraindications
- Precautions
Adult
1-2 mg/kg/d PO
Pediatric
4-8 mg/kg/d PO for severe, life-threatening bleeding with platelet counts <50,000/mm3, or for all patients with platelet counts <30,000/mm3
- Dosing
- Interactions
- Contraindications
- Precautions
Estrogens may decrease clearance; concurrent use with digoxin may cause digitalis toxicity secondary to hypokalemia; phenobarbital, phenytoin, and rifampin may increase metabolism (consider increasing maintenance dose); monitor for hypokalemia with coadministration of diuretics
- Dosing
- Interactions
- Contraindications
- Precautions
Documented hypersensitivity; viral, fungal, or tubercular skin infections
- Dosing
- Interactions
- Contraindications
- Precautions
Pregnancy
B – Fetal risk not confirmed in studies in humans but has been shown in some studies in animals
Precautions
May cause severe infections, hyperglycemia, edema, osteonecrosis, myopathy, peptic ulcer disease, hypokalemia, osteoporosis, euphoria, psychosis, myasthenia gravis, and growth suppression; abrupt discontinuation may cause adrenal crisis
Methylprednisolone (Solu-Medrol, Depo-Medrol)
Decreases inflammation by suppressing migration of polymorphonuclear leukocytes and reversing increased permeability. Used as alternative glucocorticoid of choice for all patients with severe, life-threatening bleeding or children with platelet counts <30,000/mm3. Careful observation without medical treatment may be appropriate in some asymptomatic children.
- Dosing
- Interactions
- Contraindications
- Precautions
Adult
Loading dose: 125-250 mg IV
Maintenance dose: 0.5-1 mg/kg/dose IV q6h for up to 5 d
Pediatric
Loading dose: 2 mg/kg IV
Maintenance dose: 0.5-1 mg/kg/dose IV q6h for up to 5 d
- Dosing
- Interactions
- Contraindications
- Precautions
Coadministration with digoxin may increase digitalis toxicity secondary to hypokalemia; estrogens may increase levels; phenobarbital, phenytoin, and rifampin may decrease levels (adjust dose); monitor patients for hypokalemia when taking concurrent diuretics
- Dosing
- Interactions
- Contraindications
- Precautions
Documented hypersensitivity; viral, fungal, or tubercular skin infections
- Dosing
- Interactions
- Contraindications
- Precautions
Pregnancy
C – Fetal risk revealed in studies in animals but not established or not studied in humans; may use if benefits outweigh risk to fetus
Precautions
Hyperglycemia, edema, osteonecrosis, peptic ulcer disease, hypokalemia, osteoporosis, euphoria, psychosis, growth suppression, myopathy, and infections are possible complications
Blood products
Administration of IVIg may temporarily increase platelet counts in some children and adults with ITP. Consider IVIg if the situation requires a rapid, temporary rise in platelet count.
Intravenous immune globulin (IVIg)
DOC for severe, life-threatening bleeding or for children with platelet counts <20,000/mm3 with minor purpura; can be used alone or in addition to glucocorticoid therapy.
- Dosing
- Interactions
- Contraindications
- Precautions
Adult
1-2 g/kg IV administered over 1-5 d
Pediatric
1 g/kg once
- Dosing
- Interactions
- Contraindications
- Precautions
None reported
- Dosing
- Interactions
- Contraindications
- Precautions
Documented hypersensitivity; IgA deficiency and anti-IgE/IgG antibodies
- Dosing
- Interactions
- Contraindications
- Precautions
Pregnancy
C – Fetal risk revealed in studies in animals but not established or not studied in humans; may use if benefits outweigh risk to fetus
Precautions
Check serum IgA before IVIg (use IgA-depleted product, eg, Gammagard S/D); may increase serum viscosity and thromboembolic events; may increase risk of migraine attacks, aseptic meningitis (10%), urticaria, pruritus, or petechiae (2-30 d postinfusion)
Increases risk of renal tubular necrosis in elderly patients and in patients with diabetes, volume depletion, or preexisting kidney disease; changes in lab findings associated with infusions include elevated antiviral or antibacterial antibody titers for 1 mo, 6-fold increase in ESR for 2-3 wk, and apparent hyponatremia
Thrombopoietic Agent
These agents directly stimulates bone marrow platelet production.6
Eltrombopag (Promacta)
Oral thrombopoietin (TPO) receptor agonist. Interacts with transmembrane domain of human TPO receptor and induces megakaryocyte proliferation and differentiation from bone marrow progenitor cells. Indicated for thrombocytopenia associated with chronic idiopathic thrombocytopenic purpura in patients experiencing inadequate response to corticosteroids, immunoglobulins, or splenectomy. Not for use to normalize platelet counts but used when clinical condition increases bleeding risk.
Prescribers must enroll in Promacta Cares program. Only available through restricted distribution program. Program phone number is (877) 9-PROMACTA (877-977-6622).
- Dosing
- Interactions
- Contraindications
- Precautions
Adult
50 mg PO qd 1 h ac or 2 h pc
East Asian ancestry or moderate-to-severe hepatic insufficiency: 25 mg PO qd
Use lowest dose to achieve and maintain platelet count ≥50 X 109/L to reduce risk of bleeding; not to exceed 75 mg/d; discontinue if platelet count not increased after 4 wk at maximum dose or if platelet count increases substantially
Pediatric
Not established
- Dosing
- Interactions
- Contraindications
- Precautions
CYP1A2, CYP2C8, UGT1A1, and UGT1A3 substrate; OATP1B1 inhibitor; UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7, and UGT2B15 inhibitor
Coadministration with moderate or strong CYP1A2 (eg, ciprofloxacin, fluvoxamine) or CYP2C8 (eg, gemfibrozil, trimethoprim) inhibitors may inhibit eltrombopag’s oxidative metabolism and increase toxicity
Coadministration with UGT1A1 or UGT1A3 inhibitors or inducers may affect glucuronidation of eltrombopag
Inhibits OATP1B1 and may increase exposure to OATP1B1 substrates (eg, benzylpenicillin, atorvastatin, fluvastatin, pravastatin, rosuvastatin, methotrexate, nateglinide, repaglinide, rifampin)
Inhibits UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7, and UGT2B15 enzymes and therefore may increase systemic exposure of substrates (eg, acetaminophen, narcotic, NSAID)
Chelates polyvalent cations; allow 4-h interval for administration of other medications, calcium-rich foods, or supplements containing polyvalent cations (eg, antacids, aluminum, calcium, iron, magnesium, selenium, zinc)
- Dosing
- Interactions
- Contraindications
- Precautions
None known
- Dosing
- Interactions
- Contraindications
- Precautions
Pregnancy
C – Fetal risk revealed in studies in animals but not established or not studied in humans; may use if benefits outweigh risk to fetus
Precautions
May cause hepatic impairment, monitor ALT, AST, and bilirubin, and discontinue if levels increase; may cause bone marrow fibrosis because of reticulin fiber deposition; excessive dose may increase platelet counts and produce thrombotic/thromboembolic complications (discontinue if platelet count >400 X 109/L after 2 wk at lowest dose); may increase risk for hematological malignancies; monitor CBC count weekly during dose adjustment, monthly following stable dose, and at least 4 wk after discontinuation
Romiplostim (Nplate)
An Fc-peptide fusion protein (peptibody) that increases platelet production through binding and activation of the thrombopoietin (TPO) receptor, a mechanism similar to endogenous TPO. Indicated for chronic immune (idiopathic) thrombocytopenic purpura in patients who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Only available through the Nplate NEXUS (Network of Experts Understanding and Supporting Nplate) program, a program designed to promote informed risk-benefit decisions before initiating treatment. For more information, see www.nplate.com or call (877) NPLATE1 (877-675-2831).
- Dosing
- Interactions
- Contraindications
- Precautions
Adult
1 mcg/kg (actual body weight) SC initially; adjust in increments of 1 mcg/kg SC qwk to achieve platelet count of 50 X 109/L or greater (median dose in clinical trials was 2 mcg/kg); not to exceed 10 mcg/kg/wk
If platelet count not adequate to control bleeding after 4 wk at maximum dose, discontinue and continue monitoring platelet count for 2 wk
Pediatric
<18 years: Not established