| Pityriasis rubra pilaris= النخالية الحمراء الجرابية |
|
|
Pityriasis Rubra Pilaris
EPIDEMIOLOGY Pityriasis rubra pilaris is a rare chronic disorder with an estimated incidence ranging from 1 in 5000 to 1 in 50,000 dermatology patients. The age distribution is bimodal with peak incidences in the first and fifth decades of life. The disease occurs in all races and affects the sexes equally. ETIOLOGY AND PATHOGENESIS Although an underlying dysfunction in vitamin A metabolism has been suggested as a cause, the etiology and pathogenesis of pityriasis rubra pilaris are poorly understood. The role of vitamin A deficiency remains uncertain, because attempts to produce keratotic lesions by vitamin A deprivation have been unsuccessful. Moreover, a deficiency of retinol-binding protein as an underlying pathogenic mechanism resulting in inadequate transport of vitamin A to the skin has yet to be ascertained. Larregue et al. reported that a previous upper respiratory tract episode appeared to trigger the onset of the disease.2 A report of an acute exanthematous form of juvenile pityriasis rubra pilaris that followed an upper respiratory tract infection and initially resembled Kawasaki disease supports the hypothesis
CLINICAL FINDINGS
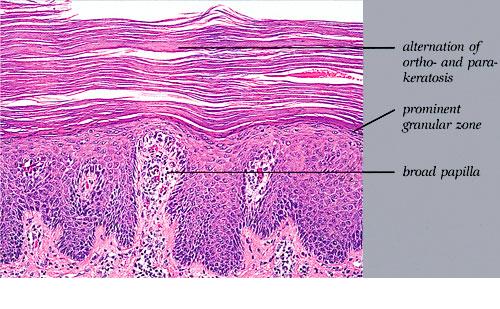
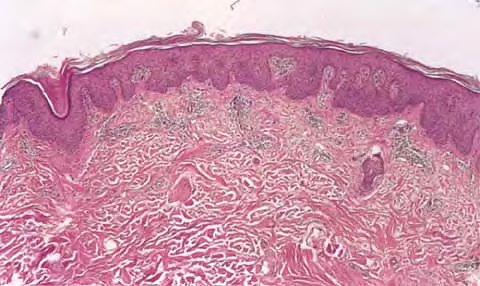
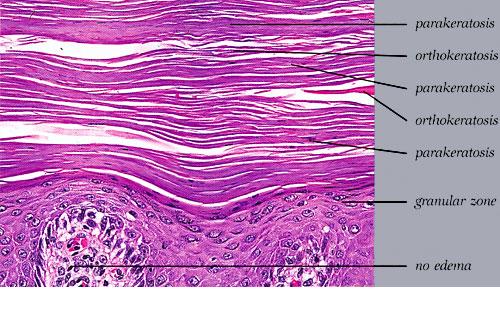
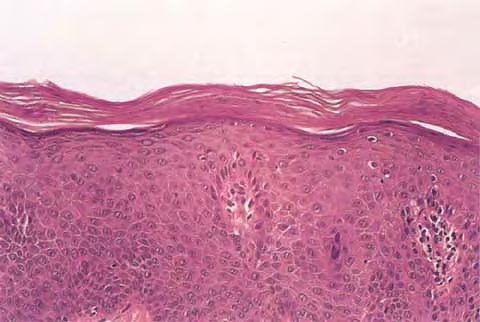
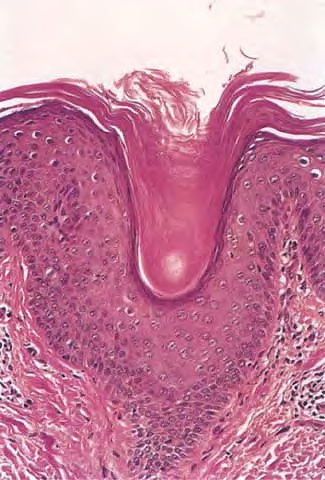
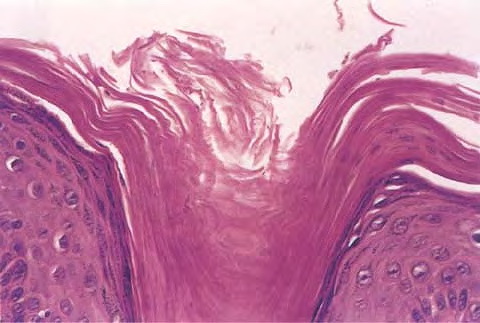
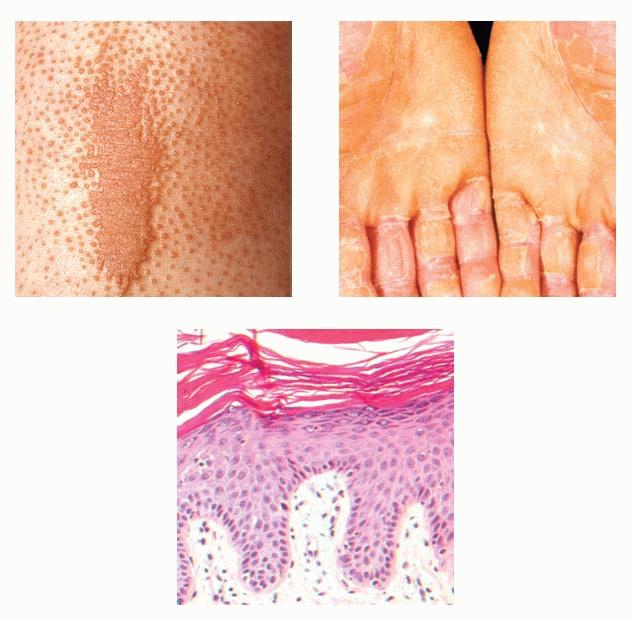
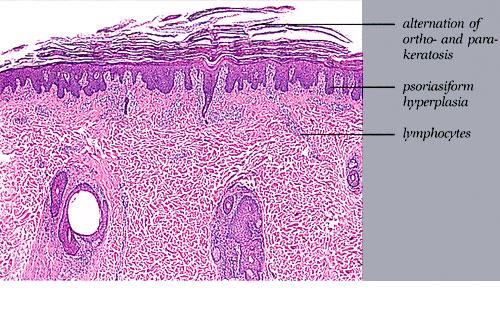
Cutaneous Lesions Pityriasis rubra pilaris is generally believed to comprise more than a single entity, and a classification scheme based on clinical characteristics and course has been proposed by Griffiths. Type I (classic adult) is the most common subtype, accounting for more than 50 percent of all cases. Characteristically, patients show an eruption of follicular hyperkeratotic papules that spread in a cephalocaudal direction . As the disease further evolves, a reddish orange, scaling dermatitis appears that often progresses to a generalized erythroderma over a period of 2 to 3 months . A diagnostic hallmark of pityriasis rubra pilaris is sharply demarcated islands of unaffected skin (“nappes claires”) in a random distribution . Many patients develop a waxy, diffuse, yellowish keratoderma of the palms and soles . Peripheral edema is common. Nail changes are not uncommon and include distal yellow-brown discoloration, nail plate thickening, splinter hemorrhages, and subungual hyperkeratosis. Eventually, the mucous membranes may be affected with a diffuse whitish appearance of the buccal mucosa as well as lacy white plaques and erosions. Hair and teeth are normal. Type II is an atypical variant with onset in adulthood. Areas of follicular hyperkeratosis as well as ichthyosiform scaling, especially on the legs, dominate the clinical picture. This variant lacks the typical cephalocaudal progression observed in type I, and there is less tendency for patients to become erythrodermic. Sparseness of the scalp hair is occasionally seen. Type III (classic juvenile) typically begins in year 1 or 2 of life and shows all the morphologic features of type I . Type IV (circumscribed juvenile) affects approximately 25 percent of patients. This type usually presents several years after birth and is characterized by well-demarcated hyperkeratotic erythematous plaques on the elbows and knees, resembling localized psoriasis. According to Griffiths,1 these lesions do not progress to the widespread types I and III. Yet, some cases show marked palmoplantar keratoderma. Type V is an atypical variant of juvenile pityriasis rubra pilaris that usually presents in the first few years of life and has a more chronic course. This type is distinguished by follicular hyperkeratosis with only minimal erythema and a scleroderma-like appearance of the hands and feet. Most cases of familial pityriasis rubra pilaris belong to this type, which may even represent a different clinical entity sharing features with several poorly defined ichthyotic disorders such as follicular ichthyosis and the erythrokeratodermas. Some reports have described a type VI variant associated with human immunodeficiency virus (HIV) infection.4 The clinical features of this variant are similar to those of type I but with increased severity and additional manifestations of acne conglobata, hidradenitis suppurativa, and lichen spinulosus. Related Findings There have been rare cases of a pityriasis rubra pilaris-like eruption, clinically and histologically, in patients with dermatomyositis, often associated with internal neoplasia. Concomitant rheumatologic disorders, mainly inflammatory polyarthritis, have also been reported. In addition, numerous other non-cutaneous diseases have been considered to be related to pityriasis rubra pilaris, which in all cases probably occurred fortuitously. PATHOLOGY Pathologic findings in pityriasis rubra pilaris vary according to the duration of the disease. The findings are most likely to be diagnostic in the acute phase, when hyperkeratosis, acanthosis with broad short rete ridges, and alternating orthokeratosis and parakeratosis oriented in both horizontal and vertical directions can be observed (see Pityriasis Rubra Pilaris At a Glance). Usually, there is a sparse superficial, perivascular lymphocytic infiltrate in the underlying dermis. Keratinous plugs of the follicular infundibula as well as perifollicular areas of parakeratosis may also be present. A prominent granular layer and dilated, but not tortuous, capillaries are features that help to distinguish pityriasis rubra pilaris from psoriasis, the most important differential diagnosis. COMPLICATIONS Systemic symptoms are uncommon except when generalized erythroderma occurs, and then they are comparable to those seen in exfoliative dermatitis. Occasionally, a mild ectropion may develop when the face becomes uniformly erythematous. Although they are rare, moderate to severe pruritus or burning sensations may occur. ▪ PROGNOSIS AND CLINICAL COURSE The classic adult disease (type I) usually remits completely within an average of 3 years . Recurrences are recognized in up to 20 percent of patients, however, sometimes after long periods of subclinical disease. In the classic juvenile variant (type III), spontaneous clearing is commonly observed in 1 to 2 years. The atypical variants (type II and IV), however, have a less favorable prognosis for remission, although some cases of type IV improve in the late teens. There is little or no tendency for type V to resolve spontaneously; improvement with administration of systemic retinoids has been described, but relapses occurred when treatment was withdrawn. Clinical manifestations in the HIV infection-associated type VI are severe and occasionally fatal, with death occurring due to complications of cutaneous sepsis. TREATMENT Patients with pityriasis rubra pilaris are often unresponsive to multiple therapies, both topical and systemic.5 Because of the relative rarity of the disease and its variable course, the chances for
Currently, oral retinoids are the first line of therapy in patients with pityriasis rubra pilaris. Isotretinoin has been reported to be of value, although a comprehensive review suggests that acitretin may be more effective in clearing lesions. Accordingly, most patients are treated first with acitretin today. Therapy with methotrexate, using the guidelines established for psoriasis has shown variable rates of success. Some cases respond well to photochemotherapy (psoralen with ultraviolet A phototherapy), some may flare, and others require combination treatment with retinoids or methotrexate. In patients with severe symptoms, effective amelioration of the disease may require extracorporeal photopheresis. Immunosuppressive agents are of inconsistent benefit. Thus, controversy persists about the role of cyclosporine in the treatment of pityriasis rubra pilaris. Although most studies show lack of efficacy, several cases of adult-type pityriasis rubra pilaris showed significant clearance in 2 to 4 weeks with this therapy. Some patients are helped by azathioprine, but this effect is also inconsistent. The use of glucocorticosteroids in the management of pityriasis rubra pilaris has been studied, but there is no evidence to suggest a beneficial effect. Whether phototherapy can be effective is controversial. Ultraviolet B irradiation, which is efficiently used for psoriasis, has not been helpful or has even worsened pityriasis rubra pilaris. Ultraviolet A1 phototherapy may be a satisfactory alternative. When conventional treatment strategies fail, new therapeutic approaches may include the use of immunomodulatory drugs. Fumaric acid esters were reported as successful in inducing remission in a patient with juvenile pityriasis rubra pilaris unresponsive to the usual treatment options.7 Moreover, there is some evidence indicating that blockade of tumor necrosis factor-α, which is beneficial in psoriasis and psoriatic arthritis, is effective in the adult-onset type I of pityriasis rubra pilaris.8 Differential Diagnosis of Pityriasis Rubra Pilaris Most Likely
Consider
Always Rule Out
Topical treatment with keratolytics (when possible, under an occlusive plastic dressing) has proved a valuable mode of adjuvant therapy in pityriasis rubra pilaris. For symptomatic relief, emollients and antihistamines provide significant benefit. Topical therapy with calcipotriol showed encouraging results. Its disadvantage is that total body treatment for the erythrodermic patient can be toxic. Success has been reported with topical aminonicotinamide 1 percent, although it is not commonly used. Pityriasis rubra pilaris associated with HIV infection has responded to triple antiretroviral therapy.
PREVENTION Pityriasis rubra pilaris is a rare but socially and psychologically disabling condition, occurring in children and adults of both sexes. Suicide remains a risk in patients with generalized disease. Knowledge of the clinical pattern and cutaneous findings, therefore, is crucial to permit early therapeutic intervention and may allow prevention of protracted illness and serious complications in this clinically challenging condition. Treatments for Pityriasis Rubra Pilaris
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||