Kaposi Sarcoma
In 1872, the Austro-Hungarian dermatologist Moriz Kaposi described five patients with multicentric cutaneous and extra-cutaneous neoplasm that primarily affected older individuals.1 This disease, originally described as idiopathic multiple pigment sarcoma, was later eponymously designated Kaposi sarcoma (KS). Since then, different variants of this tumor have been described, which have distinct epidemiologic features and run distinct clinical courses but show comparable histopathologies.
ETIOLOGY AND PATHOGENESIS
Human Herpesvirus-8 and Kaposi Sarcoma
Several decades of epidemiologic data and electron microscopic studies have suggested the involvement of an infectious agent in the etiology of KS.2 This suspicion was substantiated in 1994 when Chang and colleague discovered DNA of an unknown virus in KS lesions. This discovery led to cloning, isolation, and characterization of a novel human herpesvirus now referred to as Kaposi sarcoma-associated herpesvirus (KSHV) or alternatively, human herpesvirus (HHV)-8.
HHV-8 is a member of the γ-herpesviridae subfamily, genus rhadinovirus. Its 165-kb DNA genome has been entirely sequenced and shown to contain approximately 90 open reading frames. Many of the HHV-8 regulatory genes are homologous to genes of other rhadinoviruses, and several are homologous to human genes involved in regulation of apoptosis, cell proliferation, and angiogenesis. As in other herpesviruses, HHV-8 gene expression is tightly controlled and either associated with viral latency such as LANA-1, V cyclin and vFLIP or lytic viral replication such as vGPCR, vIL 6, and v-bcl-2. Several HHV-8 gene products, both of the lytic and the latent viral life cycles, have been shown to transform cells in vitro as well as in animal models in vivo. It is therefore generally accepted that HHV-8 is a transforming herpesvirus and that this capacity plays a direct role in the development of KS.
The host range of HHV-8 in vivo and in vitro comprises vascular and lymphatic endothelial cells as well as different types of hemopoietic cells.5 The widely expressed 12-transmembrane light chain of the human cystine/glutamate exchange transporter system xc- has recently been put forward as fusion entry receptor of HHV-8. Other surface molecules such as α3β1 integrin and heparan sulphate proposed earlier to allow for HHV-8 infection appear to play only a secondary role. However, because peripheral blood CD19+ B cells, which are permissive for infection in vitro, appear not to express the cystine transporter xCT other receptor molecules are likely to be identified in the future.
The routes of HHV-8 transmission are not completely known. The high number of KS in homosexual and bisexual men and the association of tumor development with certain sexual practices early on suggested the sexual transmission
of the virus. In later epidemiologic studies sexual activities were confirmed as a route for HHV-8 transmission and it was demonstrated that the transmission risk increases with the increase of numbers of sexual partners16 and participation in certain forms of oral/genital and oral/anal sex.16,17 By contrast, in areas where the virus is widespread, such as in Mediterranean countries and in Africa, strong evidence exists that nonsexual transmission among family members16 plays a major role. The oropharynx is the site of strongest viral replication and high HHV-8 copy numbers are present in saliva.18 Based on these findings, the nonsexual transmission via saliva is widely assumed to play the most important role in childhood transmission in endemic areas.19 Vertical transmission from mother to child during pregnancy or birth seems not to play an important role for the spread of the virus.16 For the medical profession, it is important to remember that HHV-8 may also be transmitted by blood and blood products as well as from donor organs in the course of transplantations.
HHV-8 is present in all KS variants and the evidence for a causal relationship between HHV-8 infection and KS tumor development is overwhelming. Among the most important arguments for a role of HHV-8 in KS pathogenesis are the findings that (1) the HHV-8 viral genome is detectable in KS lesions of all stages regardless of the clinical variant; (2) within KS lesions HHV-8 is present in latent form in virtually all tumor cells; and (3) KS tumor cells show monoclonal integration of viral DNA.Furthermore, the prevalence of HHV-8 infection is largely congruent with that of KS: In areas of low incidence of KS, such as the United States and Northern Europe, HHV-8 infection is very rare (below 0.1 percent).16,25 By contrast, in areas where high incidence of KS has been reported, such as in Southern Italy, HHV-8 prevalence is much higher and approaches 20 percent. As expected, the highest HHV-8 prevalence has been found in central Africa in the regions known to be hotspots of African endemic KS. In this region, the seroprevalence in adults ranges from 22 percent to 71 percent. Similarly, in human immunodeficiency virus (HIV)-1-infected patients in developed countries, HHV-8 prevalence is highest in homosexual and bisexual men and reflects the incidence of KS in this risk group.
The time span from infection with HHV-8 to development of KS depends on the clinical type. Immunosuppression/dysregulation plays an important aggravating role and leads to disease manifestation in organ transplant recipients within 1 to 2 years and in HIV-1 infected patients 5 to 10 years after infection.16 For classic KS this time span is far longer and the contributions of either host or environmental factors are not yet established.
In addition to KS, HHV-8 has been implicated in the pathogenesis of pleural effusion lymphoma, a rare B cell neoplasm seen primarily in acquired immunodeficiency syndrome (AIDS) patients, and multicentric Castleman disease (MCD). In the former, HHV-8 is invariably present and diagnostic, in the latter it is found in the majority (> 95 percent) of HIV-infected patients with MCD but only in approximately 40 percent of HIV-negative MCD patients.10 MCD has been associated in the past also with classic KS.
Histogenesis
The histogenetic derivation of the KS tumor cells (i.e., the cells lining the irregular vascular-like spaces of early and angiomatous lesions and the cells composing the bundles and interlacing fascicles of solid tumors) has been the subject of intense research over the past two decades. The immunophenotype of these cells (i.e., vWF+/-, PAL-E-, CD31+, CD34-, VEGFR3+) characterizes them as part of the lymphatic endothelial differentiation lineage.29,30 The recent demonstration that HHV-8 replicates better in lymphatic than in blood vascular endothelial cells is a further argument for this assumption.31 However, the finding that infection of blood vascular endothelial cells with HHV-8 leads to a phenotypic switch towards lymphatic endothelial cells31,32 leaves the possibility that an immature angioblast either resident in the skin or circulating and able to differentiate into both endothelial cell lineages is the initial precursor of KS tumor cells.33,34
Some controversy also surrounds the question of whether KS is a reversible proliferative disorder or a true malignant neoplasm. Analyses of the methylation pattern of the human androgen receptor gene have shown that lesions with both clonal and polyclonal proliferations can be identified.5,35,36 Moreover, multiple lesions that arise in an individual patient may develop independently from distinct transformed cells and later acquire clonal characteristics.5 Studies of HHV-8 terminal repeat sequences which show monoclonality in nodular KS lesions support these findings37 and indicate that KS starts as a polyclonal disease with subsequent evolution to a monoclonal process.
Classic Kaposi Sarcoma
EPIDEMIOLOGY
The annual incidence rates for classic KS vary greatly in different geographic regions and ranges from 0.14 per million inhabitants (for both women and men) in Great Britain to 10.5 per million in men and 2.7 per million in women in Italy.38 A preponderance of male patients has existed since the original description and a male-female ratio of approximately 3:1 is found in large surveys of most regions.38,39 The higher KS incidence in countries surrounding the Mediterranean has led some authors to speak of a Mediterranean KS variant as a distinct entity. However, because the clinical course is comparable to that of classic KS, Mediterranean KS is not treated separately here.
CLINICAL MANIFESTATIONS
The typical patient with classic KS is a Caucasian male in his sixties with a Mediterranean or Jewish background. The tumors usually start on the skin of the distal portions of the lower extremities as unilateral or bilateral bluish-red (hematoma-like) macules. These lesions tend to progress slowly both horizontally and vertically and develop into firm plaques (Fig. 128-1) and, eventually, into nodules. During tumor progression the color changes to brownish and the skin overlying the tumor becomes hyperkeratotic and, in particular on the lower extremities, may ulcerate. Surrounding the tumor there is frequently a pitting edema which may evolve into fibrosis. Classic KS usually runs a rather benign course slowly extending proximally and patients may sometimes live with slowly progressive disease for decades.38,39,41 After several years of progression (Fig. 128-2), dissemination to other body sites is frequently seen, and tumors may become manifest in lymph nodes, on mucous membranes, and in internal organs such as the gastrointestinal tract where it is rarely symptomatic.41,38 Because classic KS affects mostly elderly patients, death from other causes may precede its full spread.
African Endemic Kaposi Sarcoma
EPIDEMIOLOGY
An increased frequency of KS in black Africans in Central Africa was noted several decades before the outbreak of the AIDS pandemic. In some countries of this region, KS accounts for up to 10 percent of all malignancies in men. A prevalence of male patients, with male-female ratios ranging from 3:1 in children and up to 18:1 in adults is seen with endemic African KS. The age at onset is lower than that in classic KS at 35 to 39 years for males and 25 to 39 years for females.
CLINICAL MANIFESTATIONS
African-endemic KS occurs as nodular, florid, infiltrative, and lymphadenopathic types. The florid/vegetating and infiltrative variants are characterized by a more aggressive biologic behavior and lesions extend deeply into the dermis, subcutis, muscle, and bone. Lymphadenopathic African KS predominantly affects children and young adults and may follow a fulminant course with rapid progression.47 Skin and mucosa are affected to a lesser degree. The reasons for the different courses observed in African KS are not clear and potential co-factors have not been unequivocally identified.
Kaposi Sarcoma in Therapeutically Immunosuppressed Patients
EPIDEMIOLOGY
During the 1970s, a high incidence of KS was described in organtransplant recipients receiving immunosuppressive therapy (up to 500-fold increased as compared to the control population).48,49 Transplantation-associated KS is observed predominantly in kidney allograft recipients, whereas it occurs rarely in recipients of other solid organs and bone marrow allografts.48, 49, 50 In addition to post-transplantation, KS has been reported in patients receiving immunosuppressive therapy for other reasons, most notably the treatment of autoimmune diseases.
The clinical appearance and the course of KS in therapeutically immunosuppressed patients may be slowly progressive as with the classical variant but can also be rapidly disseminating like in AIDS KS (see the section Clinical Manifestations). KS can occur from 1 month to more than 10 years after transplantation.54 The dose and type of the immunosuppressive drug are very important risk factors for developing KS and determining its clinical course.40,50 For example the risk of KS associated with cyclosporin A is higher than with other drugs, such as glucocorticoids and azathioprine, and the disease onset is earlier.49,50 Regression of KS can be achieved by reduction or withdrawal of the immunosuppression (see the section Clinical Manifestations). Conversely, rapid tumor progression can occur after dosage increase.49,50 In transplantationassociated KS, a preponderance of male patients, with a ratio of approximately 3:1 is observed50 and in geographic regions with a high incidence of classic KS as well as in ethnic groups with a higher risk KS development, the risk for transplantation associated KS is increased.
CLINICAL MANIFESTATIONS
Skin lesions of KS in this group of patients are identical to those of classic and HIV-associated KS. In a large series, skin involvement was found in more than 85 percent of patients with transplantation-associated KS whereas less than 15 percent had visceral disease (gastrointestinal, lungs, lymph nodes) without skin involvement.
AIDS-Associated Kaposi Sarcoma
EPIDEMIOLOGY
A major harbinger of the AIDS pandemic were clusters of young homosexual men suffering from KS in large U.S. cities in 1981. During the first decade of the AIDS pandemic, KS had been diagnosed as the AIDS-defining disease in more than 20 percent of HIV-1-infected patients in Europe.56 Despite a decline in recent years, KS is still the most frequent AIDS-associated tumor in men who have sex with men. Other patient groups at risk for AIDS, including intravenous drug users, hemophiliacs, blood transfusion recipients, and children born to HIV-positive mothers in industrialized countries, are far less affected. The situation is different in Africa, where KS is the most frequent tumor arising in HIV-1-infected patients independent of risk groups, including children suffering from AIDS.
AIDS KS differs from classic KS by its more rapid course and its rapid multifocal dissemination: early AIDS-KS lesions appear as small oval violaceous macules that develop rapidly into plaques and small nodules. They are frequently present at multiple locations at
disease onset and have a tendency for rapid progression. In contrast to other variants of KS, the initial lesions in AIDS patients frequently develop on the face, especially on the nose, eyelids, and ears, and on the trunk, where the lesions follow the relaxed skin tension lines . If untreated, disseminated AIDS-KS lesions may coalesce to form large plaques involving large areas of the face, the trunk, or extremities and lead to functional impairments. The oral mucosa is frequently involved and represents the presenting site in 10 percent to 15 percent of AIDS KS . Involvement of the pharynx is not uncommon and may result in difficulty eating, speaking, and breathing.
Involvement of extra-cutaneous sites occurs more rapidly and more dramatically in patients with AIDS KS than in those with classic KS. In addition to the oral mucosa, KS lesions are most frequently found in lymph nodes, the gastrointestinal tract, and the lungs. Although gastrointestinal KS is usually found when cutaneous lesions are present, exclusive gastrointestinal involvement is possible. The stomach and duodenum represent predilection sites and KS lesions there can cause clinical symptoms such as bleeding and ileus. Although visible on gastroscopy, such lesions are underdiagnosed histologically because they are located in the submucosa and may escape the biopsy forceps. Pulmonary KS can cause respiratory symptoms such as bronchospasm, coughing, and progressive respiratory insufficiency.62 Bronchoscopy with transbronchial biopsy is most appropriate for the diagnosis of pulmonary KS.
.
HISTOPATHOLOGY
The histopathology of KS is dependent on the stage of KS development. Early patch-like lesions exhibit rather discrete histopathologic changes, consisting mainly of an increase in the number of dermal vessels, outlined by slightly irregular endothelial cells (Fig. 128-5). These vessels, located mainly in the superficial dermis are parallel to the skin surface, are frequently slightly irregular, and may form bizarre slits and clefts. In the surrounding skin focal hemosiderin, deposits and extravasated erythrocytes can be found as well as a moderate inflammatory infiltrate. Important differential diagnoses of this stage include lymphangioma and granulation tissue.
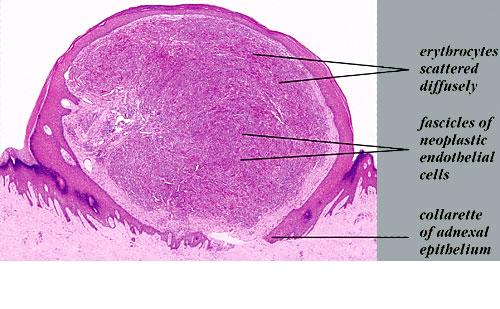
The pathology of KS plaques is more characteristic and reveals extensive vascular proliferation at all levels of the dermis with multiple dilated and angulated vascular spaces dissecting the collagen leaving a spongy network of collagen tissue. A characteristic sign of KS papules is the presence of solid cords and fascicles of spindle cells arranged between the jagged vascular channels. This biphasic angiomatous and solid tumor morphology changes to a clear-cut sarcomatous morphology with progression of the disease.
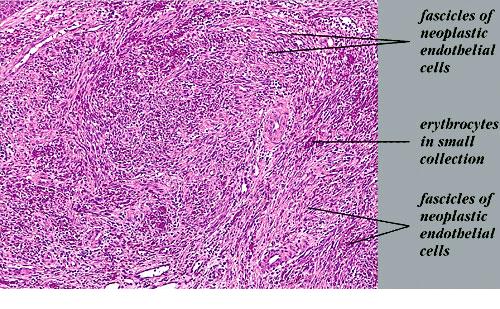
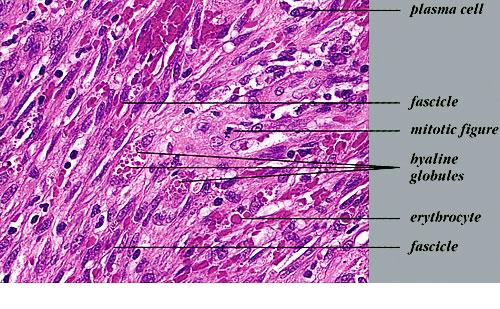
Nodular lesions consist predominantly of spindle cells arranged in bundles and interlacing fascicles and interspersed, irregular, slit-like vascular spaces without endothelial linings. Advanced lesions may display pronounced pleomorphism, nuclear atypia, and mitotic figures. At the periphery of solid tumors, (lymph) angiomatous-like portions of KS with bizarre vascular lumina and intravascular and extravasated erythrocytes, as well as siderophages, may be preserved. Erythrocytes, which appear as eosinophilic globules, are trapped within the slits and clefts formed by the spindle cells and erythrophagocytosis are occasionally observed. As in all other stages of KS, a moderate inflammatory infiltrate consisting of lymphocytes, histiocytes, plasma cells, and, sporadically, neutrophils is regularly present.
Clinical Differential Diagnoses of Cutaneous and Mucosal Kaposi Sarcoma
Most Likely
- · Localized early disease
- o Hematoma
- o Stasis dermatitis
- o Bacillary angiomatosis
- o Pyogenic granuloma
- o Well-differentiated angiosarcoma
- o Acroangiodermatitis (pseudo Kaposi)
- o Bacillary angiomatosis
- o Non-Hodgkin lymphoma
- o Squamous cell carcinoma
- o Bacillary angiomatosis
Consider
- o Dermatofibroma
- o Melanoma
- o Dermatofibrosarcoma protuberans
- o Leiomyosarcoma
- o Kaposiform hemangioendothelioma (in children)
- o Blue rubber bleb nevus syndrome
- o Metastasizing melanoma
- o Cutaneous metastasis of internal malignancies and leukemias
- o Erythema elevatum et diutinum
- o Angiosarcoma
Always Rule Out
- · HIV-1 infection
- · Non-HIV-1-related causes of immunosuppression
- · Melanoma
HIV = human immunodeficiency virus.
▪ TREATMENT
The treatment modalities for KS depend on the clinical type, the extent of the lesions,
and on the organ system involved . Despite the overwhelming evidence that infection with an oncogenic herpesvirus, i.e., HHV-8 plays a central role in KS pathogenesis; anti-herpetic drugs have so far been only of experimental value.67 Because AIDS KS incidence has been much higher than that of the other clinical variants in the past decades and due to the fact that KS in AIDS patients quickly progress into a life-threatening disease, studies on AIDS KS have brought about many of the recent insights and advances in the treatment of KS.
Localized Cutaneous Disease
Regardless of the clinical variant, if a patient presents with only limited disease confined to the skin, local therapies aimed at eliminating individual lesions are frequently used as initial treatment. Although recurrence rates are high, local therapies may satisfactorily slow progression of classic KS for several years. By contrast, in the other clinical variants it is imperative that local therapy is supported by additional measures such as change of the immune suppressive therapy in transplantationassociated KS and initiation of highly active antiretroviral therapy (HAART) in patients with AIDS KS.
Local therapies include surgical excision, local destruction with liquid nitrogen, laser or photodynamic therapy, and topical therapy with 9-cis retinoic acid. Radiation therapy is useful in localized disease at sites that are difficult to reach by other local approaches such as the nose and oral mucosa. It should also be considered in more advanced KS, in particular in resource poor settings.
PROGRESSIVE CUTANEOUS DISEASES AND
INVOLVEMENT OF OTHER ORGANS
Classic Kaposi Sarcoma.
Chemotherapy, including doxorubicin, bleomycin, vincristine, etoposide and dacarbazine, alone or in combination has been given to patients with rapidly progressive or widespread classic KS especially if internal organs are involved. However, due to the low incidence of classic KS, large comparative prospective studies are not available. As in AIDS KS, liposomal anthracyclines are effective and in one recent retrospective study were demonstrated superior to low dose interferon-α.
Kaposi Sarcoma in Patients with Therapeutic Immunosuppression.
KS in transplant patients responds well to a reduction of immunosuppressive therapy. Recently, replacement of calcineurin inhibitors by rapamycin has led to dramatic therapeutic improvement with complete remission of advanced KS lesions. This effect seems to be due to the modulation of the immune response by the change of therapy and to direct antitumor effects of the drug. Rapamycin may therefore also be of potential benefit for treating KS in patients not receiving immunosuppressive therapy.
AIDS-Associated Kaposi Sarcoma.
Until the advent of HAART, treatment of advanced AIDS KS had been mostly palliative with either combination chemotherapies or radiation therapy. During the mid 1990s HAART became available and it became possible for the first time to achieve sustained suppression of HIV-1 replication and immune recovery. Subsequent to the introduction of HAART a decrease in AIDS KS incidence and frequent remission of early KS lesions were observed during therapy. Besides their effects on HIV-1 replication, protease inhibitors (one drug of the combination antiretroviral therapy) have been suggested to have a direct antitumor effect and trials evaluating these drugs in HIV-negative patients with KS are currently under way. However, HAART alone is not sufficient to control KS that has already advanced. Therefore, if despite successful HAART and the control of HIV-1 replication, widespread lesions and/or the involvement of organs other than the skin occurs, addition of chemotherapy is required.82 Of the chemotherapeutic regimens used in the past, liposomal anthracyclines have been shown to be more efficient and better tolerated than combination therapies such as bleomycin and vincristine or both combined with doxorubicin . A typical drug regimen consists of 20 mg/m2 liposomal anthracyclines given intravenously
every 2 to 4 weeks. An advantage of the use of liposomal anthracyclines is their continued therapeutic efficacy in patients who have been previously treated by other forms of chemotherapy. For patients with anthracycline-refractory AIDS KS, paclitaxel has recently been added as potential rescue therapy.87 Interferon-α which has been a mainstay therapeutic approach for AIDS KS during the 1980s and early 1990s still holds some promise for AIDS patients with early disseminated KS who simultaneously receive HAART.
Box 128-2 Treatment Options for Kaposi Sarcoma
Localized disease
- o Surgical excision
- o Cryotherapy
- o Topical 9-cis retinoic acid
Disseminated disease/internal organ involvement
- o For patients with AIDS—initiate highly active antiretroviral therapy
- o For patients on immunosuppressive therapy—re-evaluate drug regimen
- o For AIDS patients not responding to highly active antiretroviral therapy alone—systemic cytotoxic chemotherapy
- §
- § Paclitaxel (100 mg/m2 given every 2 wk)
- o For patients with classic Kaposi sarcoma
- §
- § Vinblastine (6 mg IV once a week)
- § Doxorubicin//bleomycin//vincristine (20-30 mg/m2//10 mg/m2//1-2 mg//every 2-4 wk)
- § Interferon-α (3 million-30 million units daily to 3×/wk)
- o Investigational treatments
- § Thalidomide
- § Vascular endothelial growth factor-antisense
- § COL-3 (matrix metalloproteinase inhibitor)
AIDS = acquired immunodeficiency syndrome.
ANGIOSARCOMA AT A GLANCE
- ·
- · Occurs in 3 clinical variants:
- o Primary angiosarcoma (mostly affects elderly with a predilection for head, neck, and face).
- o Angiosarcoma in areas of chronic lymphedema [most frequently in patients after lymph-node dissection due to mammary carcinoma— (Stewart-Treves syndrome)].
- o
- ·
- · Several very rare low-grade angiosarcomas exist:
- o Endovascular papillary angioendothelioma
- o Epithelioid hemangioendothelioma
- o Retiform hemangioendothelioma
In addition to the therapies mentioned above there are several experimental therapies ranging from vascular endothelial growth factor antisense to anti-herpetic drugs currently being evaluated in clinical trials