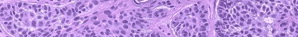
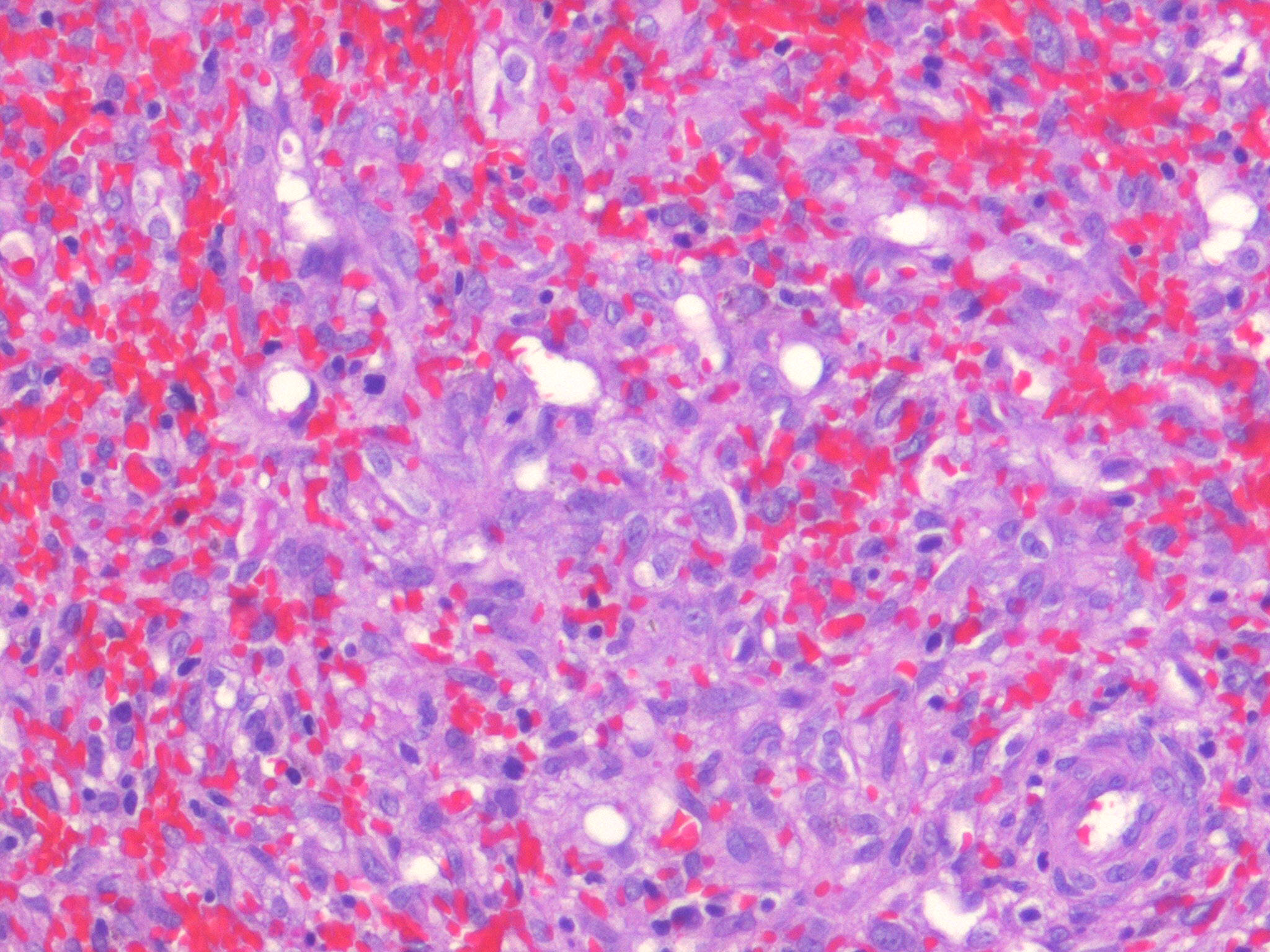
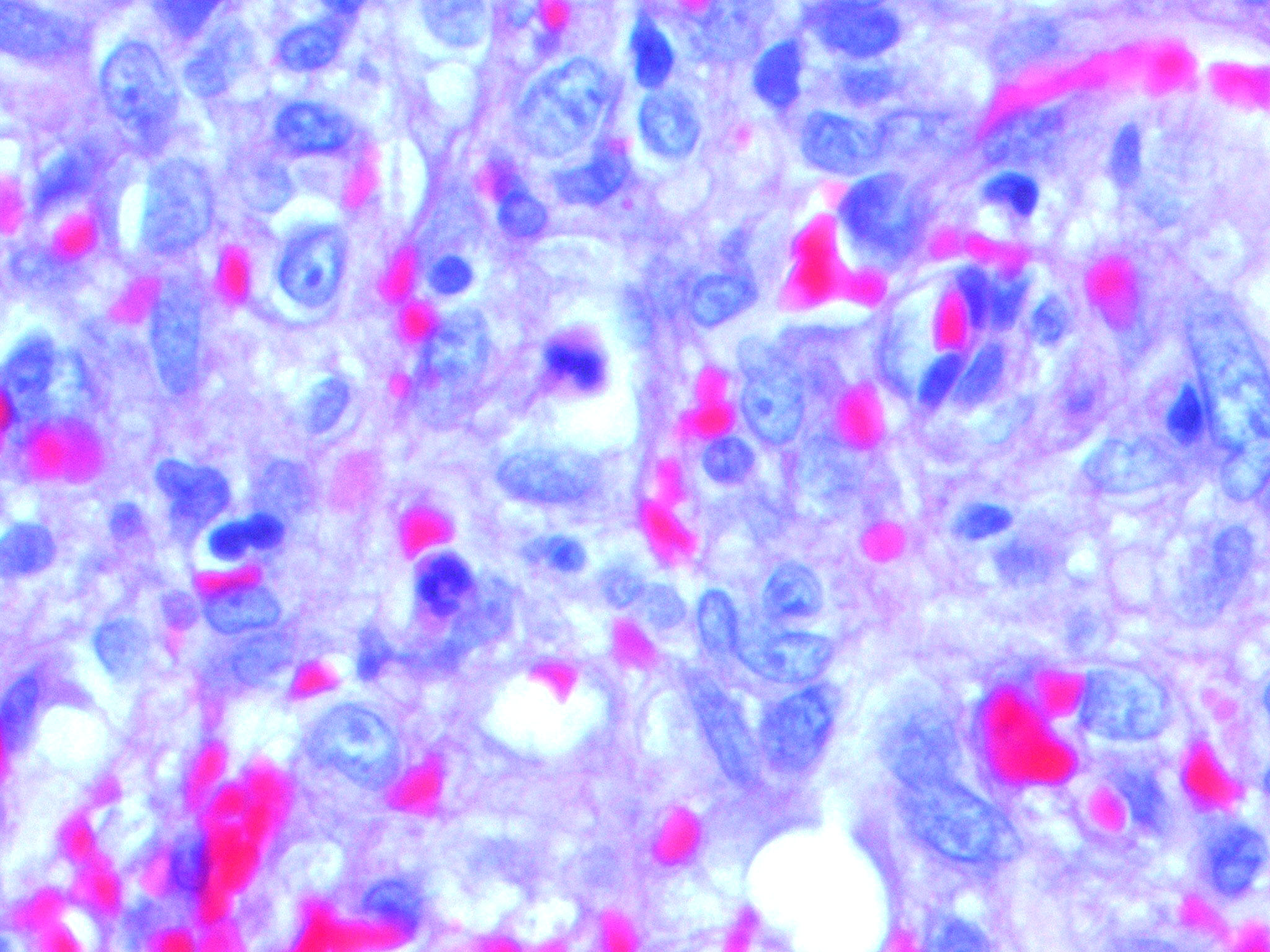
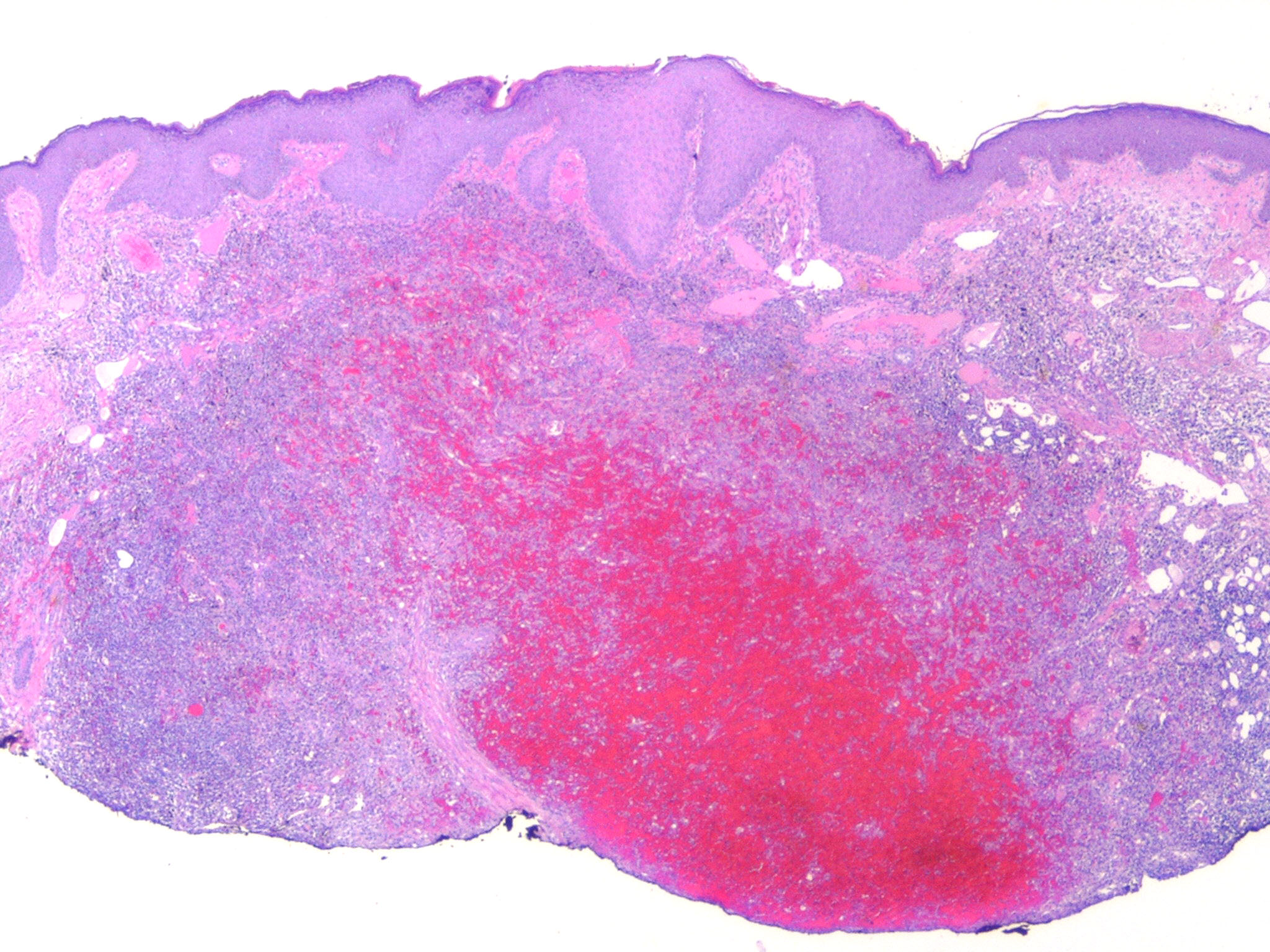
Kaposiform Hemangioendothelioma
KHE is a rare vascular tumor that has usually been reported in association with
KMP. It may be present at birth or develop in early childhood. Rare adult cases have been reported. It may present as a brown-red stain at birth which begins to thicken and become purpuric, or as a plaque or nodule similar to tufted angioma. Regional lymph node involvement may occur, but not distant metastases. Spontaneous involution is rare. Most reported cases have had associated KMP, but the KHE can occur in the absence of a coagulopathy.
The histology of KHE is characterized by spindled cells with minimal atypia and infrequent mitoses lining slit-like or crescentic vessels containing hemosiderin. The tumor is GLUT-1 negative. The overlap of both clinical and histologic features has led some to suggest that TA and KHE are part of a spectrum of disease. Increased lymphatic spaces have suggested a lymphatic origin, similar to that of Kaposi sarcoma.
Kasabach-Merritt Phenomenon
KMP has been used to describe infants with vascular tumors and thrombocytopenia with an associated coagulopathy .It was long considered to be a complication of “hemangioma,” but it is now recognized to be a complication of TA and KHE, not IH.
KMP must be differentiated from a coagulopathy which can arise in association with venous and mixed venouslymphatic malformations (sometimes erroneously called KMP) due to chronic clotting and consumption of clotting factors, but not primarily by platelet trapping. The hallmark of KMP is severe thrombocytopenia. Consumption of fibrinogen, elevated D-dimers, and decreased coagulation factors occurs to varying degrees. Most cases involve the skin and musculature, but deeper viscera including cervicothoracic, abdominal, and pelvic regions can be affected. Although the tumors causing KMP can persist, the coagulopathy usually abates by 1 year of age or sooner with treatment. KMP may resolve with atrophic or stain-like areas, infiltrated plaques and papules, or nodules. Residual fibrosis is not rare and can result in considerable morbidity.113
No one treatment is uniformly effective. Options include corticosteroids, vincristine, cyclophosphamide, actinomycin-D, methotrexate, interferon-α, ticlopidine plus aspirin, surgical excision, arterial embolization, and radiotherapy. Platelet transfusions should be avoided unless active bleeding occurs or before surgical procedures.
Multifocal Lymphangioendotheliomatosis with Thrombocytopenia
Multifocal lymphangioendotheliomatosis with thrombocytopenia (also known as cutaneovisceral angiomatosis with thrombocytopenia), a rare condition, is characterized by multiple cutaneous vascular papules and plaques that are usually present at birth with more developing over time. Affected infants have intermittent thrombocytopenia and lesions in the gastrointestinal tract, leading to gastrointestinal bleeding. Other reported sites of involvement include bones, synovium, lungs, liver, and spleen. Skin biopsy specimens demonstrate thin-walled vessels, some hobnailed endothelial cells, and intraluminal papillary projections similar to Dabska tumor.
Spindle Cell Hemangioendothelioma
Spindle cell hemangioendothelioma is a rare vascular tumor most often seen with Maffucci syndrome. It can occur at any age and site but the extremities are the most commonly affected. The histology is of a nodular, dense, spindle cell proliferation in association with dilated dysplastic veins. Lesions can be locally aggressive and may recur even after excision.
Congenital Eccrine Angiomatous Hamartoma (Sudoriparous Angioma)
Congenital eccrine angiomatous hamartoma is a rare condition characterized by ill-defined plaques with increased lanugo hair and sweating at the site of the lesion. They are usually located on the extremities or abdomen. Diagnosis is established on the basis of characteristic histologic findings: closely packed eccrine sweat glands associated with dilated capillaries, a few dysplastic venous channels, and a dense collagenous matrix.
Pyogenic Granuloma (Lobular Capillary Hemangioma)
Pyogenic granuloma (PG) is also known by its correct histopathologic description lobular capillary hemangioma. It is one of the most common vascular tumors of infants and children and can also occur in adults, particularly in pregnant women. PG usually presents as a solitary, red, rapidly growing papule or nodule, often with a subtle collarette of scale . Typical locations include the cheek or forehead but virtually any body site may be affected. They often develop an
eroded surface, with subsequent bleeding which can be profuse, resulting in the moniker the band-aid disease. In very young infants, PG is often mistaken for an infantile hemangioma.
PG does not involute spontaneously, but simple curettage with electrocautery is usually curative. Other options include excision, laser surgery (carbon dioxide or pulsed dye), and cryotherapy. Recurrence and even satellite lesions surrounding the original PG have been reported
.
Other Vascular Tumors
Targetoid hemosiderotic hemangioma is a benign lesion presenting as a violaceous papule, often surrounded by a pale rim and peripheral ecchymotic halo, which fades with time. Lesions usually present on the trunk or extremities and histologically consist of dilated vascular channels within intraluminal papillary projections dissecting into collagen bundles in the subcutis. Extravasated erythrocytes and hemosiderin are present, hence the designation.
Endovascular papillary angioendothelioma (Dabska tumor) is a dermal nodule or a diffuse swelling on the head, neck, or extremities. Dabska tumor is a low-grade angiosarcoma. Epithelioid hemangioendothelioma and retiform hemangioendothelioma
are other examples of low-grade angiosarcomas. These tumors, Kaposi sarcoma, andangiosarcoma are discussed in Chap. 128. Vascular malformations are discussed in Chap.