DERMATOFIBROSARCOMA
PROTUBERANS
Epidemiology
Dermatofibrosarcoma protuberans (DFSP) is a rare, locally aggressive dermal and subcutaneous mesenchymal neoplasm that typically occurs during the third and fourth decades of life, although it has been reported in patients from infancy to the ninth decade. DFSP accounts for less than 0.1 percent of all malignant neoplasms and represents 2 percent to 6 percent of all soft-tissue sarcomas. There does not seem to be a racial predilection for the tumor, however, the pigmented variant (Bednar's tumor) is more common in black patients. There seems to be a fairly equal distribution between male and female patients.
Etiology and Pathogenesis
The histogenesis of DFSP is uncertain: fibrohistiocytic, purely fibroblastic, and neural-related differentiation have all been hypothesized. A very sensitive, although non-specific, marker for DFSP is CD34. A distinctive cell population within normal nerves has been shown to express CD34, consistent with a possible neural-related histogenesis of DFSP. CD34 can help distinguish between DFSP and scar tissue in re-excision specimens.
At the molecular level, DFSP exhibits the reciprocal translocation t, producing a fusion of the collagen type I α 1 gene and the platelet-derived growth factor B-chain gene. Current techniques permit sensitive and specific detection of these fusion transcripts in paraffin-embedded DFSP tumor specimens by reverse transcription polymerase chain reaction. Both fibrosarcomatous transformation of DFSP and some cases of superficial fibrosarcoma have been shown to harbor these fusion transcripts, suggesting a close affinity between DFSP and fibrosarcoma.
Clinical Findings
HISTORY
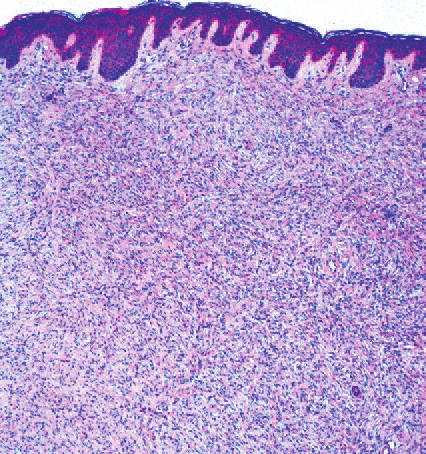
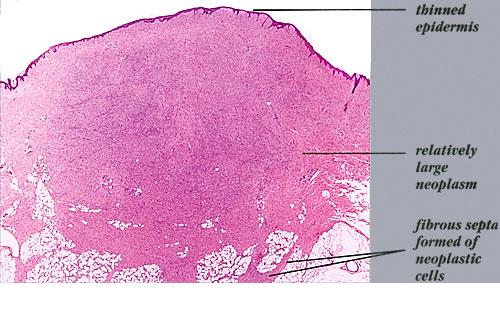
DFSP is a slow-growing lesion that often presents on the trunk and proximal extremities of patients. Due to its indolent onset, the patient may present for evaluation when the tumor is several centimeters in size (Fig. 125-1). The tumor is often misdiagnosed as a simple scar or cyst. It often grows to several centimeters in diameter and may form large, infiltrative tumors. In a small portion of patients, previous trauma at the site has been reported. DFSP has also been described as arising in scars, including surgical, burn, and vaccination sites.
CUTANEOUS LESIONS
The clinical morphology of DFSP is variable. The most common presentation is a firm, indurated plaque, often skin-colored with red-brown exophytic nodules . It also can present as a nonprotuberant, atrophic, violaceous lesion resembling morphea, sclerosing basal cell carcinoma, anetoderma, or scar .
Large nodular tumors may occur. The epidermis may be atrophic and focal ulceration may occur. The most common site of DFSP is the trunk, followed by the extremities. Approximately 15 percent of DFSP occurs on the head and neck.
MALIGNANT FIBROUS DERMAL
TUMORS AT A GLANCE
- A group of uncommon and rare cutaneous tumors with varying degrees of malignant potential.
- Includes dermatofibrosarcoma protuberans, atypical fibroxanthoma, malignant fibrous histiocytoma, fibrosarcoma, and epithelioid sarcoma.
- Diagnosis is made by biopsy.
- Dermatofibrosarcoma protuberans is a locally aggressive tumor with a high rate of local recurrence with rare metastases.
- Atypical fibroxanthoma is an intermediate-grade neoplasm that usually arises on sun-damaged skin in the eighth decade.
- Malignant fibrous histiocytoma represents a morphologic pattern that a large number of poorly differentiated neoplasms share.
- Epithelioid sarcoma is a rare tumor that classically presents on the hands and fingers of young males, which can mimic nonneoplastic inflammatory lesions.
LABORATORY TESTS
Histopathology.
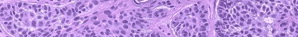
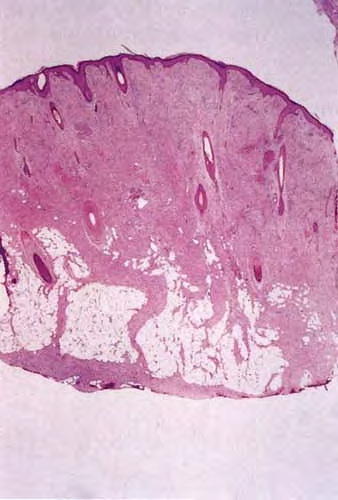
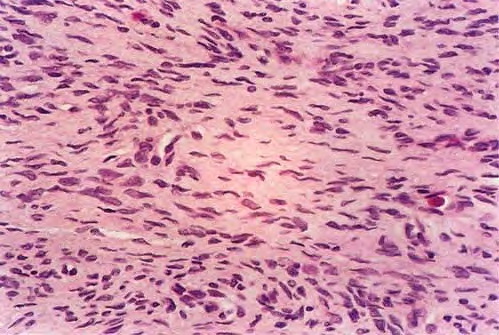
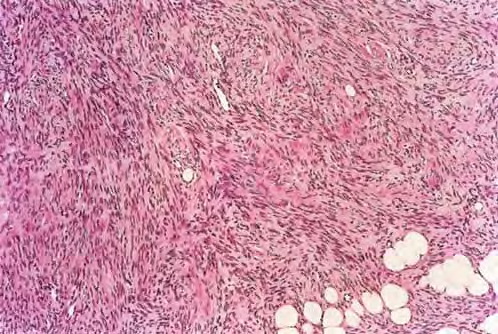
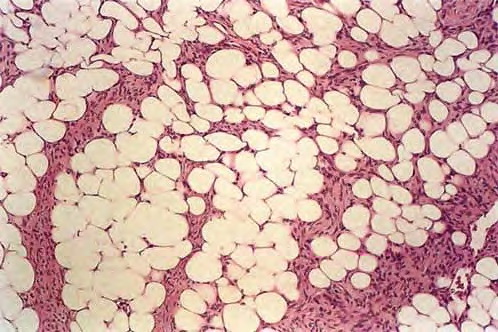
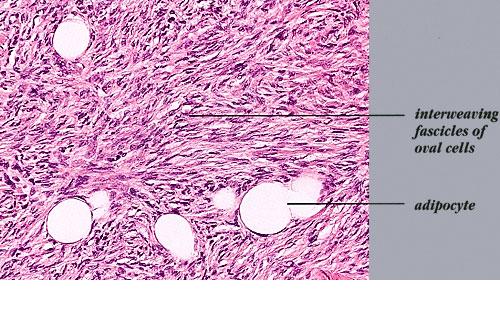
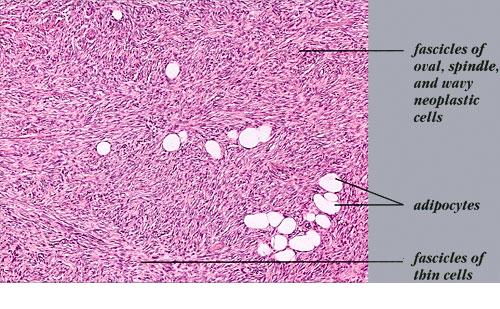
DFSP is diagnosed with a skin biopsy. A deeper wedge biopsy may be required to diagnose the atrophic variant. Histologically, DFSP is a poorly circumscribed dermal proliferation of monomorphous, slender or slightly plump spindle cells with little pleomorphism arranged in a storiform pattern . The proliferation commonly infiltrates the subcutaneous fat, isolating adipocytes to form lucencies (“honeycomb” or “Swiss cheese” pattern). In contrast to dermatofibroma, the tumor is much more cellular and usually does not have mature collagen interspersed between fascicles of spindle cells.
Variable histologic patterns described include myxoid, neuroid, fibrosarcomatous, myxoid, and granular cell types. The tumor may be relatively monomorphous or show combinations of various patterns within the original tumor or in the recurrences. Some lesions show overlap between giant cell fibroblastoma and DFSP. The fibrosarcomatous variant has basic features of DFSP but focal areas in which the cells exhibit a high grade of atypia and are arranged in long fascicles in a herringbone (fibrosarcoma-like) pattern. This transformation appears to be related to mutations in the p53 pathway. Several series indicate an increased biologic aggressiveness of this type compared with ordinary DFSP In a series from Memorial Sloan-Kettering Cancer Center, patients with this histologic variant had a 28 percent 5-year recurrence-free survival, as compared to 81 percent of those with classic DFSP. However, in a series from the Cleveland Clinic, patients with sarcomas arising in DFSP did not have an increased risk of distant metastasis within a 5-year follow-up period, providing that adequate surgical excision with wide margins was performed. It is estimated that there is a 5 percent transformation rate from DFSP to conventional fibrosarcoma. The true impact of fibrosarcomatous changes seen within a DFSP remains uncertain.
Special Tests.
Immunohistochemical staining with CD34 is useful to distinguish DFSP from dermatofibroma, which stains positively for factor XIIIa. Other markers, such as CD44, a membrane glycoprotein, and hyaluronate, a component of the extra-cellular matrix, may serve as markers to better distinguish DFSP from dermatofibroma as well. Apolipoprotein D also may have future application as a marker for DFSP.
Differential Diagnosis
Complications
DFSP is a low-grade malignant tumor, often infiltrating diffusely through the dermis and into the subcutaneous fat but seldom metastasizing. Increased age, high mitotic index, and increased cellularity are predictors of poor clinical outcome. Approximately 30 percent to 50 percent of DFSPs recur locally after simple excision, so it is generally recommended that a wide excision with 1- to 3-cm margins to the fascia or periosteum be performed (see Treatment).
Prognosis and Clinical Course
Local recurrences even after wide excision are common, although, much
greater success seems to be had with using the Mohs technique for tumor extirpation. Metastases of DFSP are extremely rare, usually occurring in the setting of multiply recurrent tumors. Metastases to the lung are most common, with nodal disease the next most common site of spread. Metastatic disease portends a poor prognosis. It has been suggested that tumors with fibrosarcomatous change may have a higher chance of recurrence or metastases.
Box 125-1 Differential Diagnoses of Malignant Dermal Tumors
Dermatofibrosarcoma protuberans
- Classic presentation
- Keloid/hypertrophic scar
- Dermatofibroma
- Fibrous histiocytoma
- Nodular melanoma (Bednar variant)
- Atrophic presentation
- Morphea
- Lichen sclerosis
- Morpheaform basal cell carcinoma
Atypical fibroxanthoma/superficial malignant fibrous histiocytoma
- Squamous cell carcinoma
- Basal cell carcinoma
- Amelanotic malignant melanoma
Epithelioid sarcoma
- Granuloma annulare
- Rheumatoid nodule
- Gouty tophi
- Dupuytren contracture
- Infectious ulcer
Treatment
Surgical removal with clear margins is the goal with DFSP. With standard excision, margins of 1 to 3 cm may be necessary to achieve clear margins. Risk of local recurrence decreases with increasing surgical margins. Pathologic examination of margins during surgery is helpful in delineating the extent of the tumor. Multiple case series have shown Mohs surgery to be an extremely effective method of resection of DFSP, with an extremely low rate of local recurrence. As a result, Mohs micrographic surgery represents the preferred surgical approach of many practitioners. Adjuvant radiation therapy may help decrease the local recurrence rate. The use of imatinib, a chemotherapeutic agent that targets the molecular translocation distinctive to this tumor, has shown promise in the setting of localized advanced disease (i.e., unresectable tumors) or metastatic disease. Imatinib may also emerge in the future as an adjunct to surgery.
GIANT CELL
FIBROBLASTOMA
Giant cell fibroblastoma, a rare neoplasm, now known to be a juvenile variant of DFSP, typically presents as a dermal or subcutaneous mass, which most frequently involves the trunk, thigh, or inguinal region. It presents much earlier in life, often within the first decade, although occurrences in adults have been reported. Histologically, this tumor has dense connective tissue with a paucity of fibroblasts and occasional wreath-like giant cells. There are also scattered mast cells present. These tumors resemble DFSP with their pattern of tumor infiltration. The tumor cells infiltrate the subcutis and surround adnexal structures. Most characteristically, the stroma displays prominent cracking artifact, with the formation of pseudovascular spaces lined by a discontinuous layer of enlarged, multinucleated tumor giant cells with coarse nuclear chromatin and scalloped nuclear borders. Immunohistochemically, these tumors stain positive for CD34 and occasionally α1 antichymotrypsin. They stain negative for factor VIII, von Willebrand's factor, CD31, actin, desmin, epithelial membrane antigen, and S100. As noted previously, this tumor shares the identical translocation with DFSP—(t17;22), collagen type I α 1 gene-platelet-derived growth factor B-chain gene.
The clinical behavior of giant cell fibroblastoma is much like that of DFSP. It shows local aggressive behavior, frequent local recurrences (up to 50 percent), and rare metastasis. Treatment is the same as for DFSP.