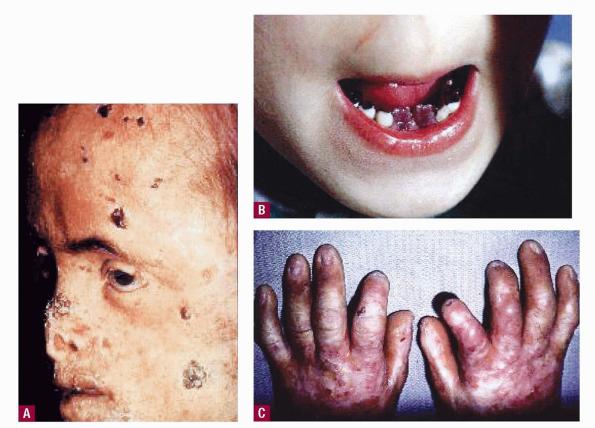
Erythropoietic porphyria (Günther disease).
congenital
Erythropoietic porphyria (EP) is a rare inborn error of porphyrin-heme synthesis inherited that is as an autosomal recessive trait. The inheritance of 2 mutant alleles for the gene encoding the enzyme uroporphyrinogen III synthase leads to accumulation of porphyrins of the isomer I type that are biologically useless but cause cutaneous photosensitivity characterized by blisters, erosions, and scarring of light-exposed skin.
Clinical manifestations can range from mild to severe. Chronic damage of skin, cartilage, and bones can cause mutilation. Hypertrichosis, erythrodontia, and reddish-colored urine are often present. Hemolytic anemia can be mild or severe, with resultant splenomegaly and osseous fragility.
The following is a selection of other eMedicine porphyria-related articles:
- Erythropoietic Protoporphyria
- Porphyria Cutanea Tarda
- Pseudoporphyria
- Variegate Porphyria
Pathophysiology
Erythropoietic porphyria is primarily a disorder of bone marrow heme synthesis. Deficient activity of the enzyme uroporphyrinogen III synthase in erythrocyte precursor cells causes a shift of the pathway away from the isomer III porphyrinogen production that can effect the end-product heme; isomer I porphyrinogens that cannot be used to form heme may be overproduced. The accumulated isomer I porphyrinogens are spontaneously oxidized to their corresponding porphyrins, which are water-soluble photosensitizers with a reddish hue.
These porphyrins are released from the maturing erythrocytes into the plasma and are excreted by renal mechanisms; urine with a port-wine color is produced. The interaction of excess porphyrins in the skin and light radiation causes photoxidative damage of biomolecular targets that is manifested as mechanical fragility and blistering that may result in severe scarring.
The hemolytic anemia of erythropoietic porphyria can cause hypersplenism in more serious cases. Hypertrophy of the bone marrow in such cases can lead to osseous fragility and pathologic fractures. Acral osteolysis and onycholysis may occur; bones and teeth are stained red by the deposition of porphyrin pigment. Ocular damage can lead to blindness. The photoactive nature of porphyrin molecules results in the bright pink fluorescence of these pigments in urine, teeth, and bones under Wood light illumination.
History
The typical complaint is blistering and fragility of light-exposed skin in an individual with discolored urine. The presentation of erythropoietic porphyria at birth in a patient with a history of a difficult perinatal course and concomitant jaundice usually indicates severe disease. Patients may have a history of hemolytic anemia before the complete diagnosis was recognized. Very early prenatal expression with nonimmune hydrops fetalis has been reported.
Physical
Findings at physical examination may include the following:
- Skin
- Photosensitivity, with formation of vesicles and bullae, occurs early in the course of the disease.
- Increased fragility and erosions can contribute to mutilation, especially on the face (eg, nose, mouth, ears) and hands.
- Hypertrichosis of the face and extremities is common.
- Oral
- The teeth have a reddish color.
- The teeth fluoresce under a Wood light due to porphyrin deposition in dentine and enamel.
- Urine
- Pink staining of the diapers in the neonatal period is common.
- This staining is due to the porphyrin pigment in the urine.
- Ocular1
- Ocular manifestations of erythropoietic porphyria include blepharitis, cicatricial ectropion, and conjunctivitis. Lagophthalmos is a major cause of light-induced ocular surface aggravation.
- Scleral findings include interpalpebral fissures and pink fluorescence of the perilimbal sclera under a Wood light.
- Subsequent bilateral corneal scarring may occur, with eventual blindness. The risk for malignant conjunctival degeneration is low.
- Skeletal
- Porphyrins are also deposited in the bone, where they cause an orange-red fluorescence.
- The severe loss of bone with subsequent contractures and deformities occurs in most adults with erythropoietic porphyria.
- X-ray studies show osteopenia and acro-osteolysis.
Causes
Erythropoietic porphyria is caused by autosomal recessive inheritance of genes that encode abnormal uroporphyrinogen III synthase enzyme protein. The resultant deficient activity of this enzyme leads to hemolytic anemia, cutaneous photosensitivity, and their complications. The mutation that causes the most severe deficiency of the enzyme uroporphyrinogen III synthase is C73R.2
The GATA gene family, a group of transcription factors, has a crucial role in normal human hematopoiesis. A mutation in GATA1, an X-linked transcription factor, has been reported in association with erythropoietic porphyria
Laboratory Studies
- Porphyrin analyses
- Urinary porphyrin concentrations are increased 100-1000 times and involve predominantly uroporphyrin I.
- Urinary excretion of uroporphyrin III and coproporphyrin III is also elevated; however, the level is less than that of the isomer I porphyrins.
- Urinary delta-aminolevulinic acid and porphobilinogen levels are not increased in erythropoietic porphyria.
- Erythrocytes most often contain increased levels of uroporphyrin I; also, elevated zinc protoporphyrin is observed in some patients.
- The combination of elevated urinary and erythrocyte isomer I porphyrin levels is specific for erythropoietic porphyria.
- Coproporphyrin preferentially accumulates as fecal porphyrin after the decarboxylation of uroporphyrin.
- Complete blood cell count
- Excessive uroporphyrins in red blood cells appear to cause fragility; therefore, a hemolytic anemia is common.
- Consequent splenomegaly and hepatomegaly are observed.
- A test to measure uroporphyrinogen III synthase activity is commercially available.
- Mutation analysis of the uroporphyrinogen III synthase gene (ie, DNA testing) is performed at porphyria research units in several countries and has become commercially available in the United States. See the American Porphyria Foundation for further information.
Other Tests
- Fluorescence microscopy of peripheral blood or bone marrow specimens
- Red porphyrin fluorescence in intact erythrocytes and erythroid precursor cells can be observed in smears of bone marrow specimens illuminated by violet or blue light against a dark-field background.
- The brilliant fluorescence of nuclei in erythrocyte precursor cells is specific for erythropoietic porphyria.
Histologic Findings
Similar dermatopathologic changes can be found in all types of porphyria with photocutaneous manifestations. The characteristic feature is a subepidermal blister with a slight superficial perivascular lymphocytic infiltrate. Blood vessels in the superficial vascular plexus have markedly thickened, hyalinized walls that contain periodic acid-Schiff (PAS)–positive, diastase-resistant glycoproteins. Papillary dermal tips often festoon into the blister cavity due to the increased rigidity of the hyalinized vessel walls.
Caterpillar bodies, which are eosinophilic linear structures in the roofs of bullae composed of basement membrane material, are described in blisters of patients with several forms of porphyria. Direct immunofluorescence tests reveal linear C3 and immunoglobulin G staining around the superficial vessels and along the dermoepidermal junction
Medical Care
- Absolute avoidance of sun exposure is crucial. The effects of topical sunscreens are less than satisfactory, but sunscreens may provide some protection if they contain physical light–reflective agents such as zinc oxide or titanium dioxide. Long ultraviolet and visible light wavelengths must be blocked by additional physical means to achieve the protection that most porphyria patients require. Sun-protective clothing should be worn. Commercially available plastic films can be affixed to home and automobile windows to filter out many of the offending wavelengths. Fluorescent lamps can be replaced by incandescent bulbs, which emit less light of porphyrin-exciting wavelengths.
- Oral beta-carotene has been used with limited benefit.5 Other oral measures that have been used include activated charcoal and cholestyramine to interrupt and prevent reabsorption of porphyrins. The large doses required of all of the oral agents often make their use somewhat impractical.
- Attempts to reduce erythropoiesis and lower circulating porphyrin levels by means of erythrocyte transfusions have been successful in reducing the expression of the disease. However, the complications of a chronic transfusion regimen are potentially severe. Severe hemolytic anemia with subsequent splenomegaly is one of the most pronounced consequences of erythropoietic porphyria. Splenectomy decreases the hemolytic anemia by increasing the lifespan of erythrocytes; however, the benefits are short lived.
- The use of oral alpha-tocopherol and ascorbic acid to quench reactive oxygen radicals has been advocated to reduce porphyrin-sensitized photodamage to skin elements and circulating erythrocytes.
- Topical lubrication of the eyes improves the dry eye symptoms and may stabilize visual function.
Surgical Care
- Bone marrow transplantation is reported to be successful; however, the long-term results are unknown. Life-threatening infectious complications limit the applicability of this therapeutic approach.6,7,8
Stem cell cord blood transplantation has also been reported successful in a few patients.9
Consultations
- A dermatologist may be consulted regarding sun avoidance measures and the treatment of secondary skin infections.
- An ophthalmologist can monitor ocular complications.
- A hematologist may be consulted to manage chronic transfusion therapy and to consider bone marrow transplantation.
- A surgeon may be consulted for splenectomy when hemolytic anemia is severe.
- An oral surgeon may be consulted for the application of dental resins to cover reddened teeth for cosmetic purposes.
Activity
- Absolute avoidance of sun exposure must be practiced.
- Sun-protective clothing, hats, and physical sunscreens should be used during daily activities.
- Avoidance of mechanical trauma is advised to lessen erosions and resultant scarring.
Medication
The goals of pharmacotherapy are to reduce morbidity and prevent complications.
Oral photoprotectants
Oral photoprotectants may prevent tissue damage due to light exposure, possibly by forming an internal light screen.
Vitamin A (Lumitene)
Exact mechanism of action not completely elucidated. Patient must be carotenemic before effects are observed. More than 1 internal light screen may be responsible for effects. May provide a limited level of photoprotection. Causes yellowing of skin (eg, carotenoderma). Photoprotection increases slowly over 4-6 weeks after treatment begins. When discontinued, skin color and benefits diminish over several weeks.
Adult
120-300 mg/d PO in divided doses
Pediatric
30-120 mg/d PO in divided doses