▪ CUTIS LAXA
(GENERALIZED
ELASTOLYSIS)
Epidemiology
Few cases reported.
Etiology and Pathogenesis
Cutis laxa (CL) describes a group of disorders that share loose, redundant skin as a feature. Most cases are inherited in an AD, autosomal recessive, or X-linked manner but acquired cases have also been described.
Because of histologic abnormalities in elastin quantity or structure in virtually all forms of CL, investigations into the molecular pathogenesis have focused on genetic mutations that result in alterations in the structure or processing of elastin in the ECM. The genetic heterogeneity of CL reflects the fact that several genes are required for normal elastin biosynthesis and function. Decreased synthetic activity of skin fibroblasts and qualitative and quantitative abnormalities of elastic fibers been reported.
Elastin gene mutations that result in abnormal tropoelastin and abnormal elastic fibers result in AD CL . Elastin is one of several matrix components responsible for the properties of reversible deformability in many tissues, including the skin, lungs, and large blood vessels. The human elastin gene (ELN) is 45 kb in length and has been mapped to 7q11.2. Elastin initially is synthesized as individual tropoelastin molecules that are aligned on a network of elastic fibers. This alignment is stabilized by the formation of intermolecular cross-links that are mediated by the copper-dependent enzyme lysyl oxidase.
CONGENITAL CUTIS LAXA AT A GLANCE
· Inheritance may be autosomal dominant [Online Mendelian Inheritance in Man , autosomal recessive , or X-linked recessive (also known as occipital horn syndrome .
· Mutations in elastin (ELN) or fibulin-5 (FBLN5) in autosomal dominant; in fibulin-5 (FBLN5, EVEC, DANCE) or fibulin-4 (FBLN4) in autosomal recessive; a copper transport adenosine triphosphatase (ATP7A) in the X-linked type.
· Cutaneous features include pendulous, inelastic skin, with an aged facies.
· Extracutaneous manifestations may include pulmonary emphysema, aortic aneurysm, pulmonary artery and valve stenosis, hernias, gastrointestinal diverticula, joint laxity, low serum ceruloplasmin, and bilateral exostoses of the occiput (or occipital horn syndrome).
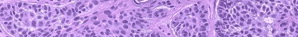
· Histopathology shows sparse and fragmented elastic fibers, better visualized with stains (i.e., Verhoeff-van Gieson or orcein).
· Information for patients and professionals at http://www.orpha.net/nestasso/cutislax/.
Type I autosomal recessive CL (OMIM #219100) is frequently fatal and is caused by mutations in the fibulin-5 gene (FBLN5, also known as EVEC or DANCE),160 which is critical for elastic fiber development.161 Mutations in FBLN5 can rarely lead to AD disease. The molecular basis for type II CL, (also known as CL with joint laxity and developmental delay; OMIM #219200),152 remains elusive, but lysyl oxidase deficiency is also found in these patients.158,162,163 A girl with CL in association with severe aortic dilatation and arachnodactyly has recently been described and found to have a mutation in the fibulin-4 (FBLN4) gene.
Mutations in the gene encoding ATP7A, a member of the P-type adenosine triphosphatase family responsible for copper transport, have been described in the X-linked type of CL, which is allelic to Menkes syndrome. ATP7A mediates copper transport from the gastrointestinal tract, efflux of excess copper from cells, and delivery of intracellular stores of copper to cuproenzymes.
Acquired forms of this disease are present after skin inflammation and structural damage of elastic fibers. Increased neutrophil elastase is found in affected skin and is presumed to disrupt elastic fiber integrity. A recent report described heterozygous mutations in FBLN5 and ELN in an adult with “acquired” CL and aortic root aneurysm after a toxocara infection; predisposition to inflammatory damage by the missense mutations is theorized.166
Clinical Manifestations
The clinical presentation of CL shows considerable heterogeneity. Congenital onset is generally indicative of an autosomal recessive or X-linked form of CL; later onset is consistent with either a heritable or acquired form. In both inherited and acquired forms, internal organs can be involved. As a rule, the AD form
of CL has primarily skin involvement, and is thus largely a cosmetic problem with a good prognosis.
The autosomal recessive and X-linked forms are often associated with more severe multisystem complications that include emphysema, diaphragmatic hernia, and gastrointestinal and genitourinary diverticula. Type I autosomal recessive CL is associated with severe cardiorespiratory complications and early death from cor pulmonale. Type II is a less severe recessive form, characterized by growth retardation, developmental delay, and ligamentous laxity. X-linked CL is characterized by mild joint laxity, bladder diverticula, hernias, and cranial occipital exostoses or horns. Many cases of acquired CL have developed after a febrile illness and/or inflammatory skin diseases such as lupus erythematous, urticaria, eczema, erythema multiforme, vesicular eruption, amyloidosis, and angioedema, as well as reactions to penicillin and D-penicillamine. CL may also be associated with amyloidosis and plasma cell dyscrasias.
CUTANEOUS FEATURES
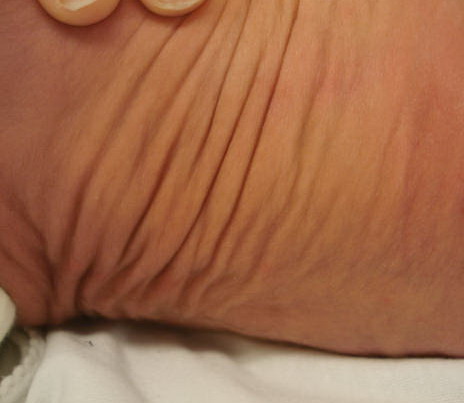
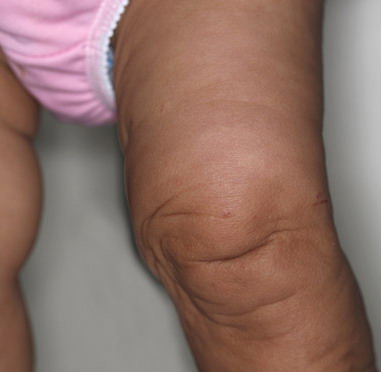
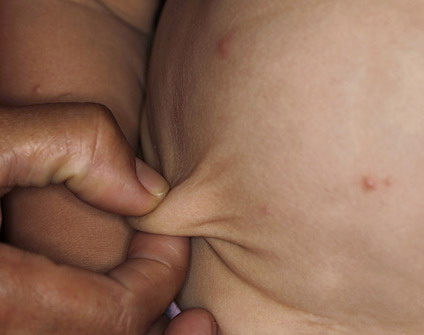
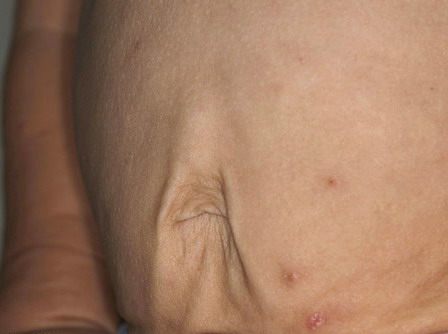
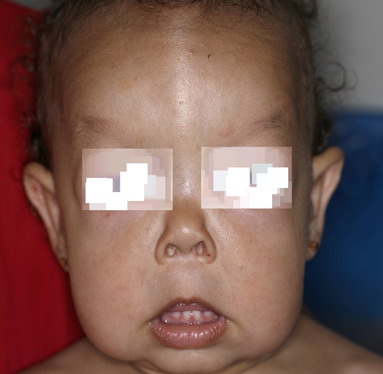
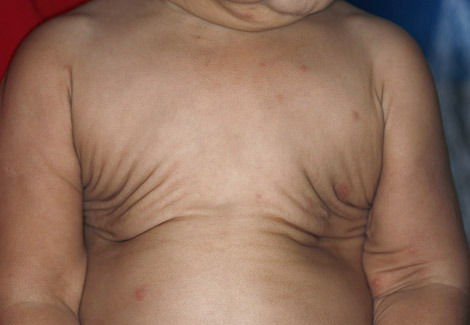
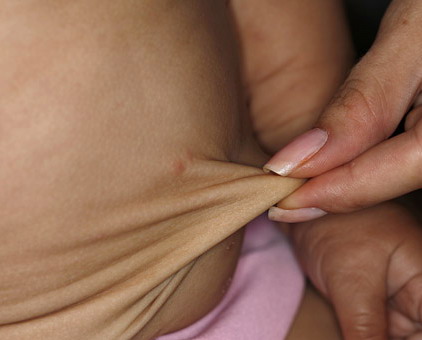
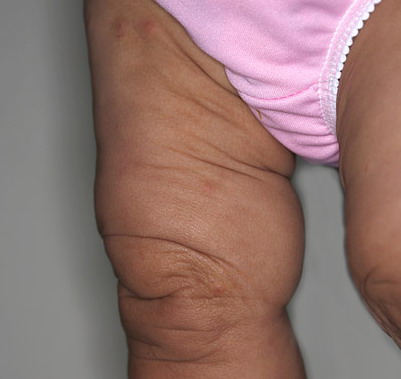
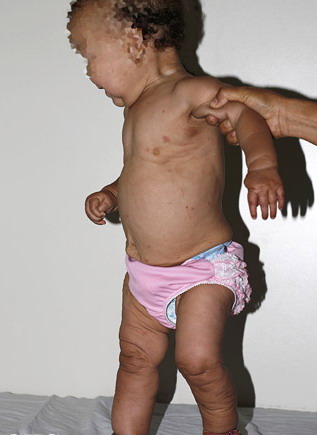
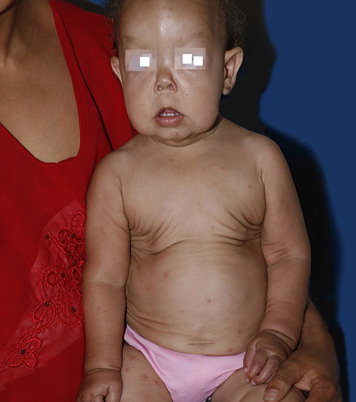
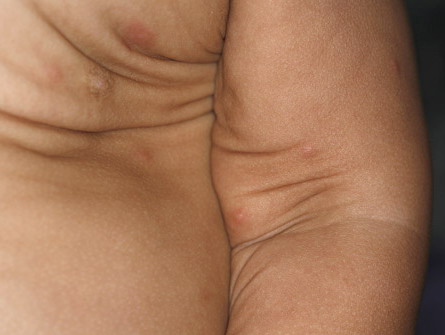
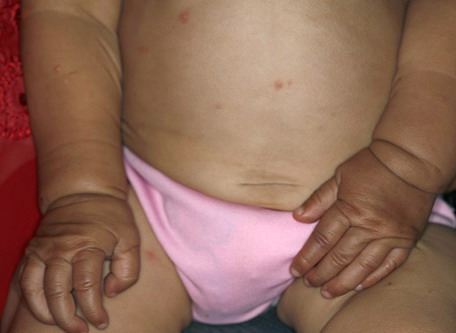
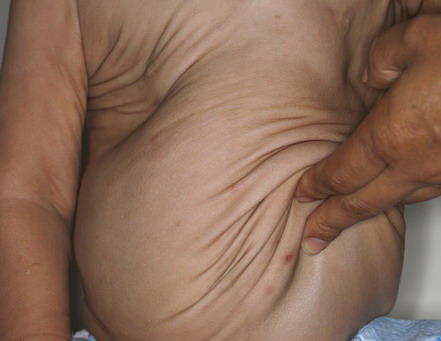
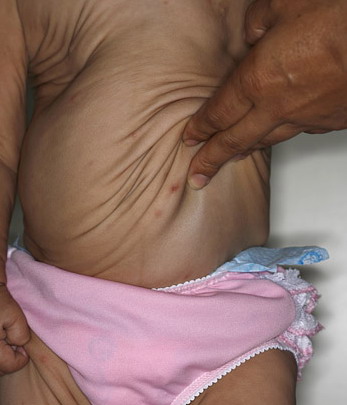
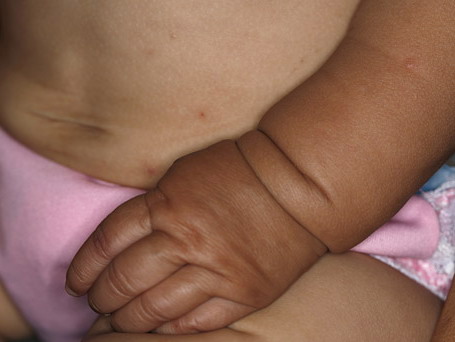
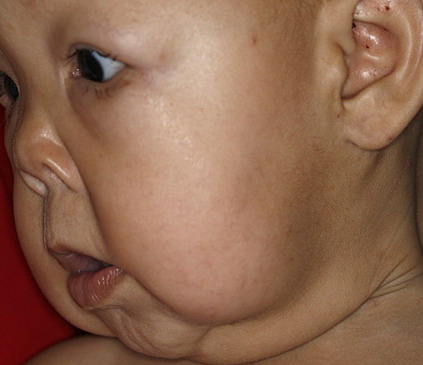
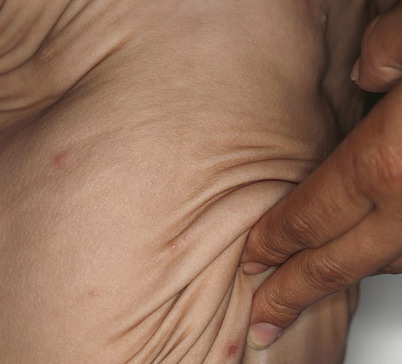
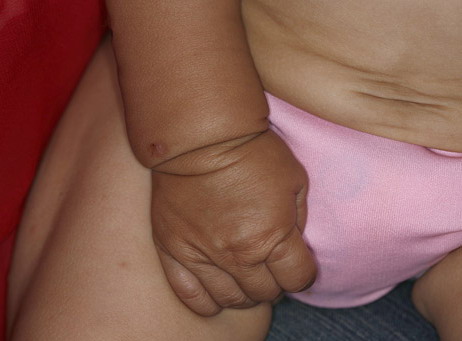
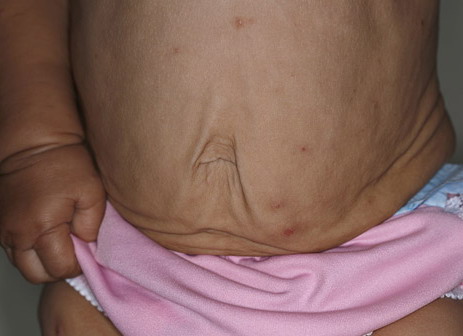
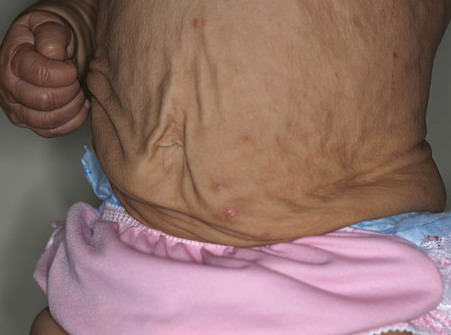
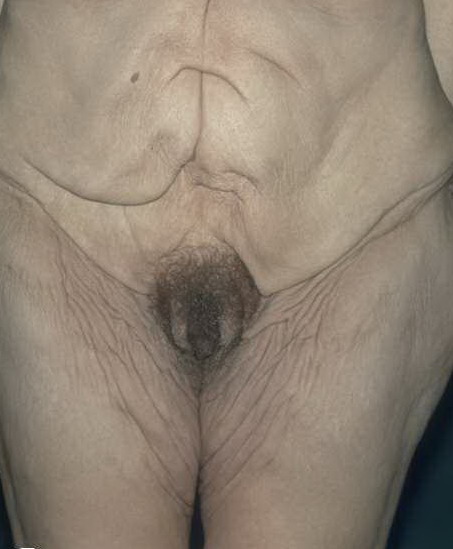
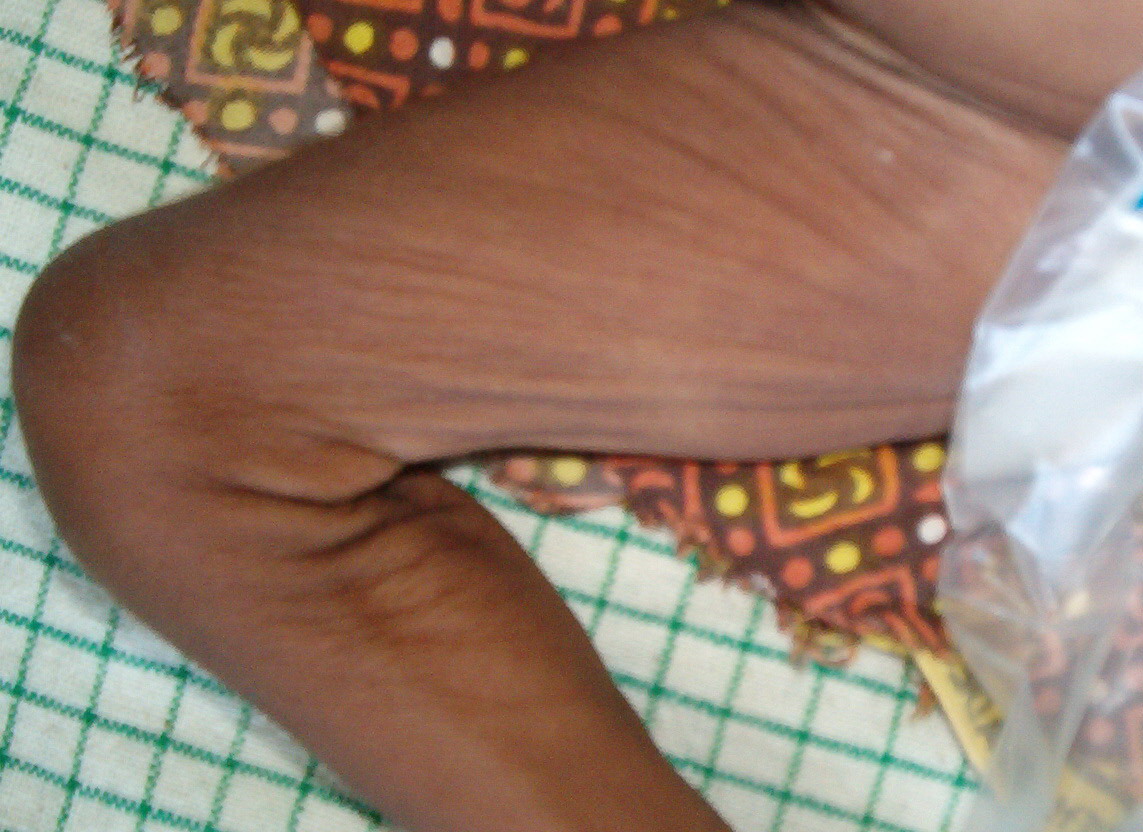
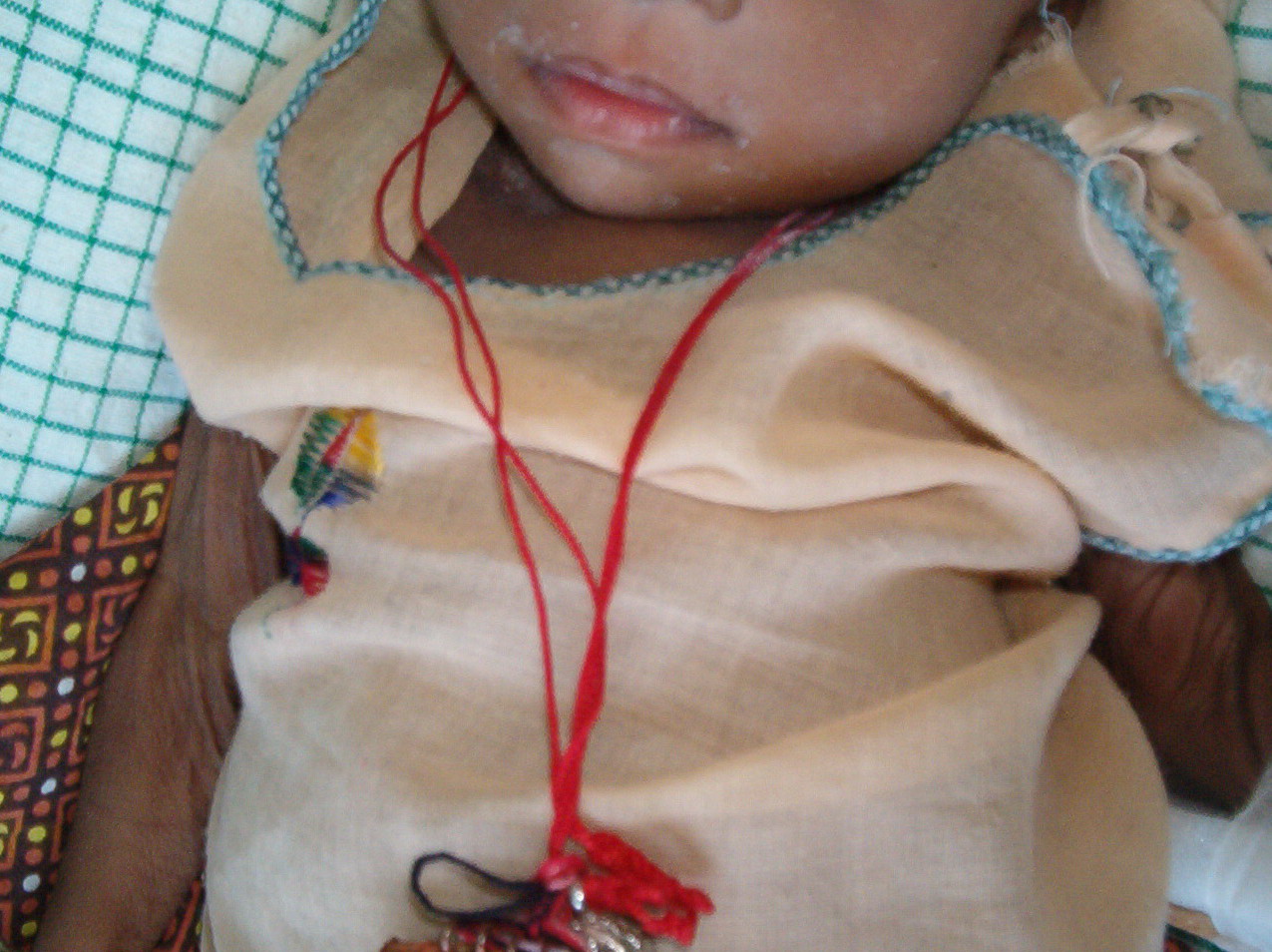
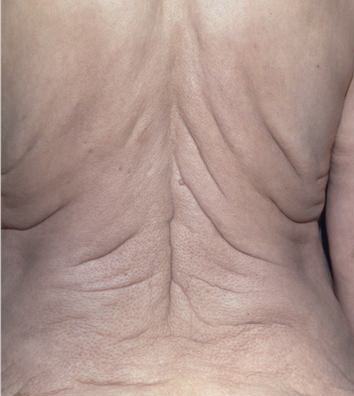
In all forms of CL, the skin gives the appearance of being too large for the rest of the body . CL may affect any portion of the body, but it often causes attention when it is present with loose appearance around the face, neck, shoulders, and thighs. At birth, the skin may be noticeably soft, loose, and hyperextensible. It tends to sag in the areas where the skin is normally loose, for example, around the face and eyes . In contrast to the skin of EDS, it returns very slowly to its normal position after being stretched; skin fragility, easy bruisability, joint hypermobility, and poor wound healing are not usually associated with CL. The skin may be excessively wrinkled and appears prematurely aged. Children with severe CL may have characteristic facial features known as the “bloodhound” look—an aged appearance with sagging jowls .
Biopsy sections of clinically affected skin show a reduced number of elastic fibers in the dermis, with the remaining fibers being fragmented, shortened, clumped, or granular. Calcifications are not present. CL can also be an acquired condition and present diffusely or localized, giving individuals the characteristic “bloodhound-like” facies.170 It is more common in adults, although children may also be affected. In more than one-half of cases, the disorder is preceded by a urticarial eruption, which may respond to dapsone, but progression will be unaffected. Common associated findings include hiatal hernias, aortic aneurysm and rupture, intestinal and genitourinary diverticula, and emphysema. Some cases with cardiovascular or pulmonary involvement may be fatal. Moreover, associated systemic conditions include multiple myeloma, lymphoma, sarcoidosis, amyloidosis, and α1-antitrypsin deficiency. Histology shows initial inflammation with concurrent loss of elastic fibers.
Differential Diagnosis of Cutis Laxa
|
SYNDROMES
|
FEATURES
|
|
DeBarsy syndrome195
|
Cutis laxa, mental retardation, cloudy corneas, athetoid posturing, dislocations, aged appearance
|
|
Lens-Majewski hyperostotic dwarfism (OMIM #151050)196
|
Skin hyperplasia, joint laxity, progeroid features, skeletal abnormalities, generalized osteosclerosis
|
|
SCARF syndrome (OMIM #312830)197
|
Skeletal abnormalities, cutis laxa, craniostenosis, ambiguous genitalia, retardation, facial abnormalities
|
|
Congenital hemolytic anemia and pulmonary emphysema (OMIM #235360)198
|
Hemolytic anemia, pulmonary emphysema, cutis laxa
|
|
Costello syndrome (faciocutaneoskeletal syndrome (OMIM #217040), mutations in the HRAS oncogene199,200 (Fig. 139-11)
|
Wrinkly skin, redundant skin of palms, soles and fingers and neck. Deep palmar creases with mildly hyperkeratotic palms and soles. Postnatal growth retardation, early feeding difficulties, mental retardation, coarse facies, thick lips and ear lobes, macroglossia, aged appearance, hyperextensible fingers and papillomas not infrequent: acanthosis nigricans, sleep apnea, cardiac abnormalities, increased risk of tumors, especially rhabdomyosarcoma, neuroblastoma199
|
|
Wrinkly skin syndrome (may be a variant of cutis laxa with growth and developmental delay) (OMIM #219200) linked to 2q32201
|
Wrinkling of skin (hands, feet, abdomen), hyperextendibility of joints (hands), dysmorphism, failure to thrive, developmental delay
|
|
OMIM = Online Mendelian Inheritance in Man; SCARF = skeletal abnormalities, cutis laxa, craniostenosis, psychomotor.
|
EXTRACUTANEOUS FEATURES
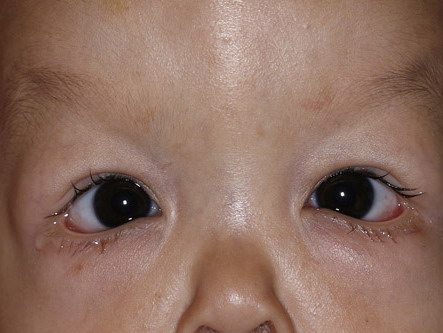
The X-linked form of CL is known as occipital horn syndrome because of occipital exostoses. Characteristic facial features include inverted lower eyelids, a hooked nose, inverted nostrils, a short columella, and a long upper lip. The cry of an affected infant may be hoarse, and a low-pitched voice is often associated redundancy of the vocal cords. Diagnostic skeletal features of this condition can be identified radiologically and include short flat clavicles, fused carpal bones, and the occipital horns, which may not be present until a few years of age. This is the only form of CL with joint laxity.
The autosomal recessive forms of CL are often associated with severe internal complications, such as genitourinary and gastrointestinal diverticula. Chronic diarrhea is a widely reported complication, and secondary kidney damage may result from diverticula causing urinary tract obstruction. The most serious complications are diaphragmatic hernia and emphysema, which may lead to cor pulmonale and death in the first few years of life, especially in patients with FBLN5 mutations. Rare cardiovascular features include aortic dilatation with secondary cardiomegaly and congestive heart failure. Rectal prolapse, diaphragmatic atony, pneumothorax, peripheral pulmonary stenosis, and aortic dilatation have also been reported. Facial features include down-slanting palpebral fissures, broad/flat nose, and large ears.
Several multisystemic disorders have features that have been called “cutis laxa,” and must be considered in the differential diagnosis listed in Box .
Treatment
Surgical correction of redundant skin folds, rectal prolapse, or any hernias is temporarily beneficial and can be undertaken without major complication. Botulinum toxin has also been used to improve facial cosmesis.173 There is no known medical treatment to prevent disease progression in the inherited
forms. Regular evaluation of internal organ involvement is important in prevention and management of major complications. Dapsone has been used to control the acute swelling of affected skin that precedes acquired CL, consistent with the suspected role of neutrophil elastase