Familial Benign Pemphigus
(Hailey-Hailey Disease)
- Familial benign pemphigus originally was described by the Hailey brothers in 1939. It is a chronic autosomal dominant disorder with incomplete penetrance. Approximately two thirds of patients have a family history of the disorder. A history of multiple relapses and remissions is characteristic. Decreased numbers of desmosomes have been implicated in the pathogenesis of benign familial pemphigus. Therapeutic options are limited.
Keratinocytes are held together through desmosomes and adherens junctions. These junctions consist of calcium-binding transmembrane glycoproteins, which contribute to cellular adhesion. Many hypotheses exist concerning the pathogenesis of familial benign pemphigus, but the cause remains uncertain. An overall defect in keratinocyte adhesion appears to be secondary to a primary defect in a calcium pump protein, ATP2C1. ATP2C1 encodes the secretory pathway Ca2+/Mn2 ATPase (hSPCA1). Mutant proteins in familial benign pemphigus create a loss of sensitivity to Ca2+ and Mn2+ ion binding and transport. Low levels of Ca2+ within Golgi bodies impair protein processing. Gene expression may be affected in benign familial pemphigus, as may phosphorylation of adhesion molecules. Localized postzygotic mutation has caused segmental manifestations of familial benign pemphigus.
No precise data are available on the incidence of familial benign pemphigus.
Familial benign pemphigus causes discomfort but is not life threatening. Benign familial pemphigus lesions often begin during the teenage years and manifest as itchy and malodorous plaques.
Both sexes are affected equally.
Familial benign pemphigus often manifests in the late teenage years or in adulthood (30s and 40s).
A family history of benign familial pemphigus usually is present. Commonly, patients may not have symptoms until ages 30-49 years. Delayed diagnosis of familial benign pemphigus also is common, especially if the patient's lesions respond to topical corticosteroids, antibiotics, or antifungals.
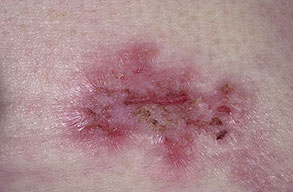
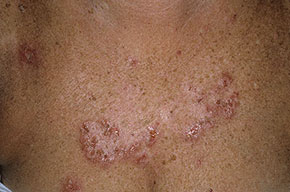
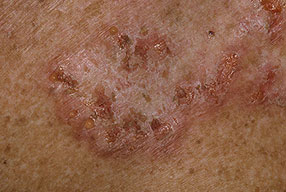
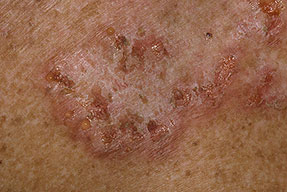
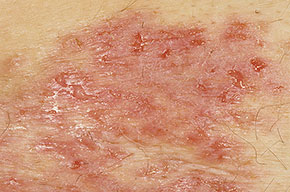
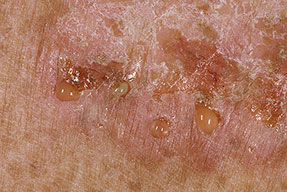
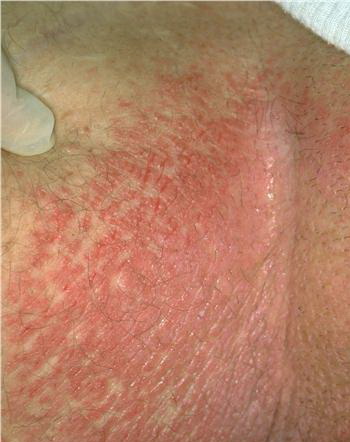
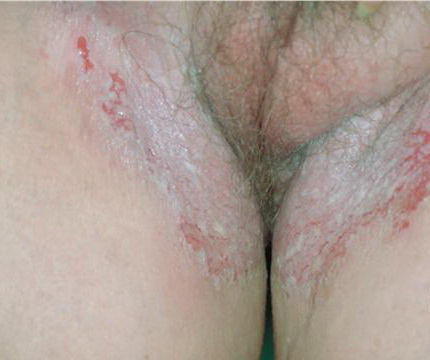
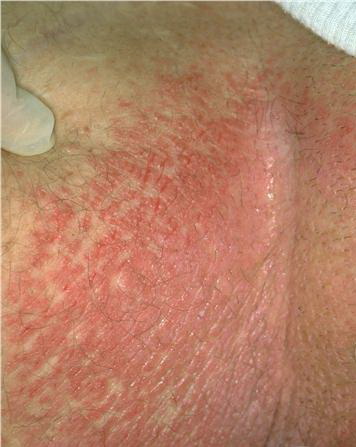
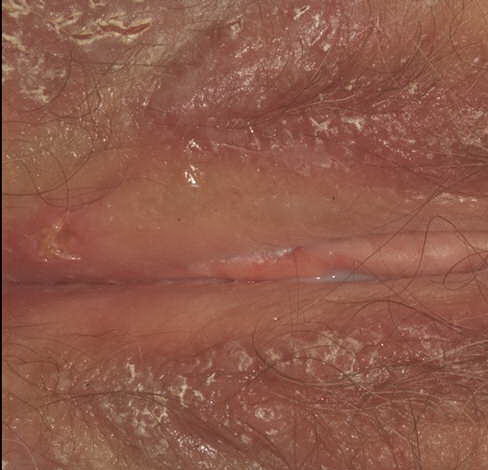
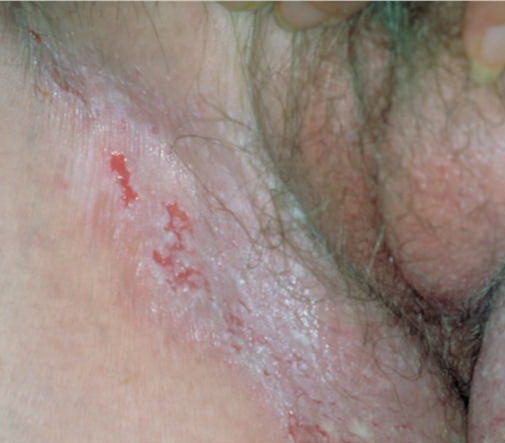
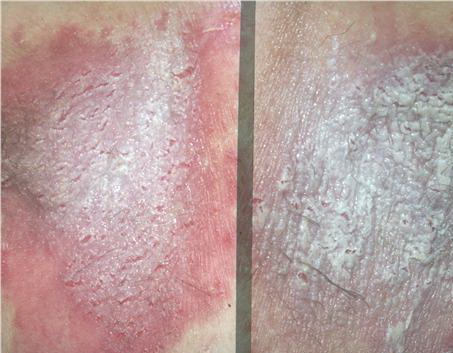
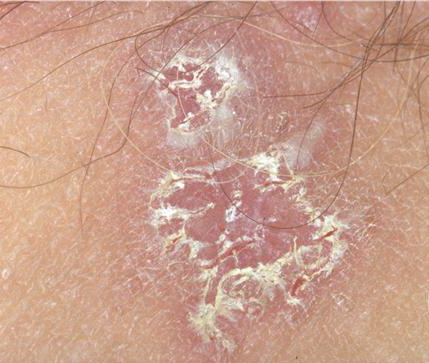
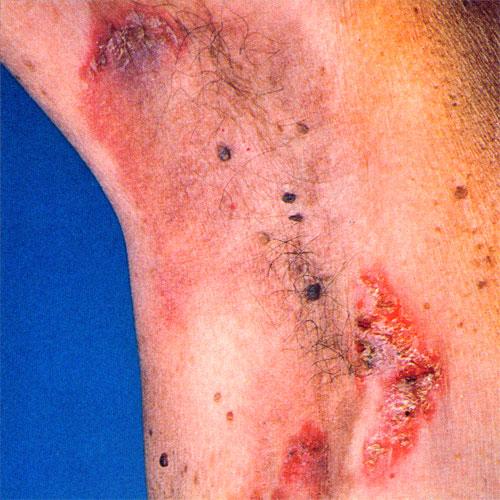
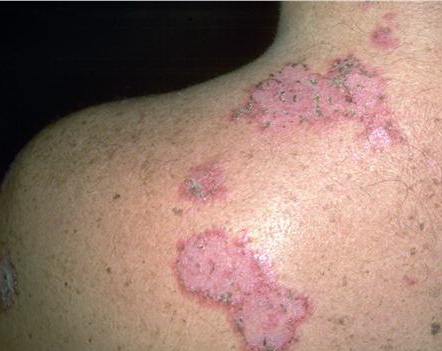
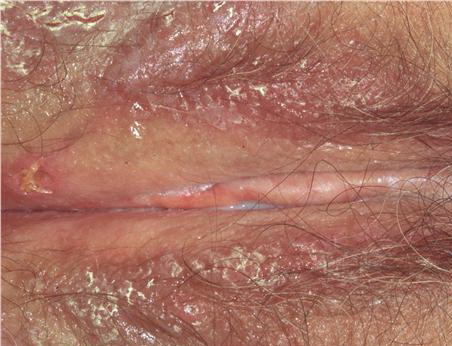
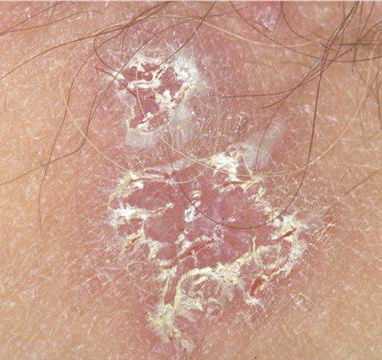
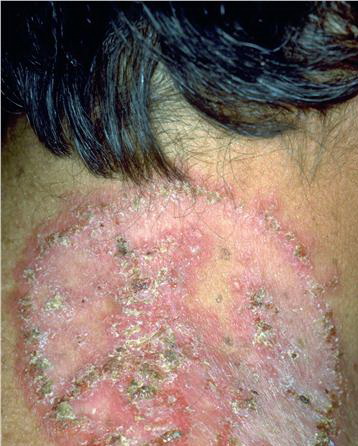
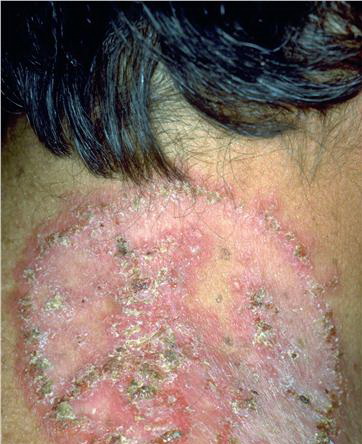
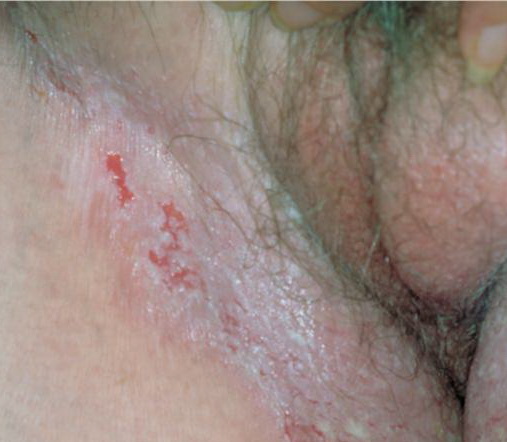
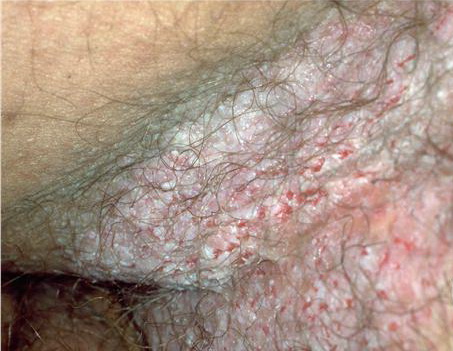
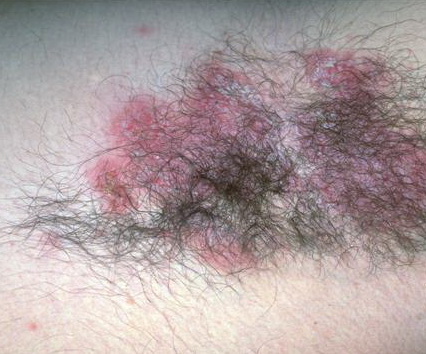
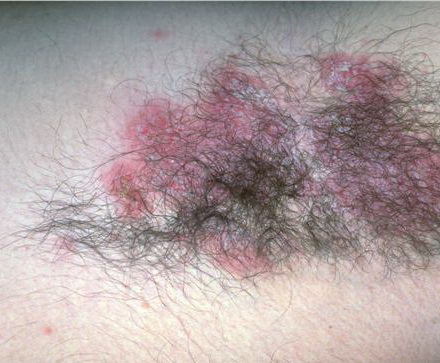
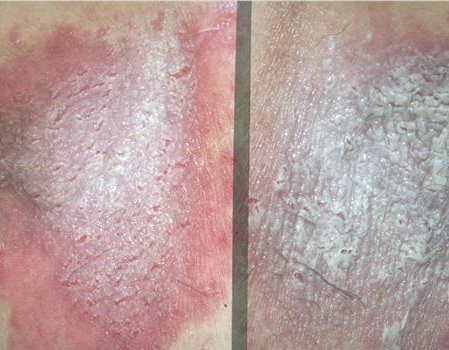
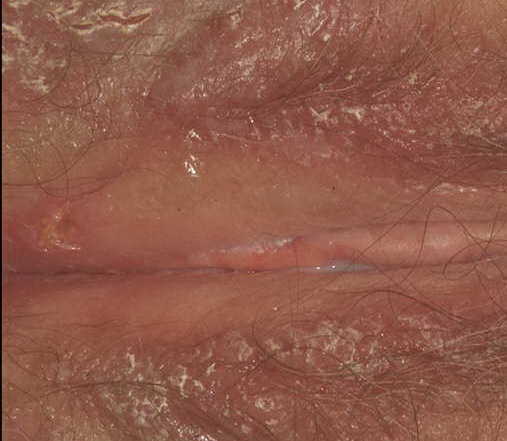
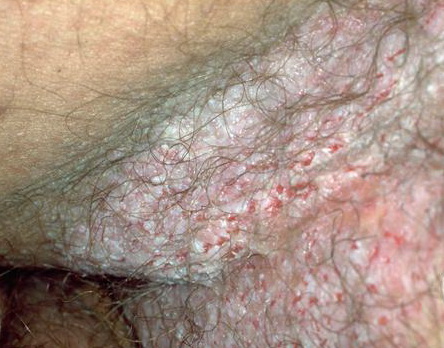
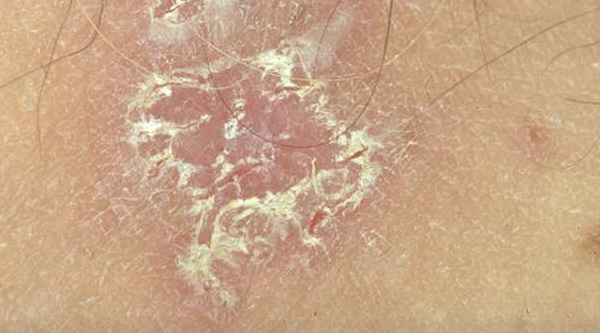
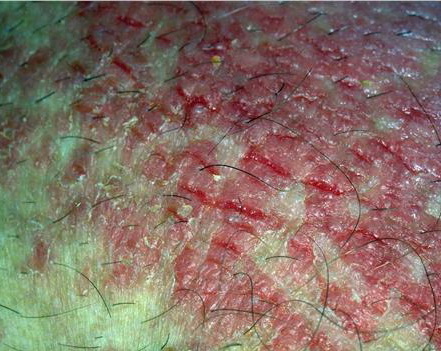
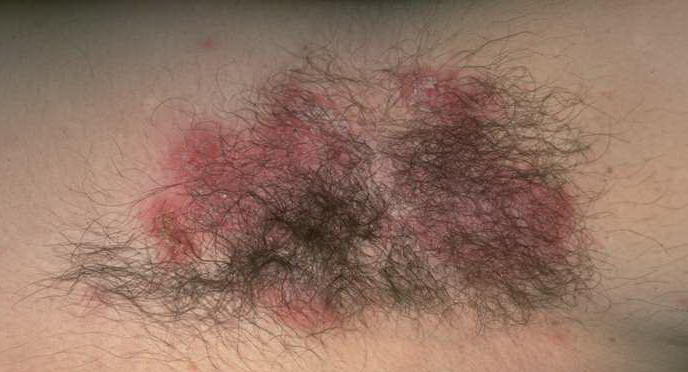
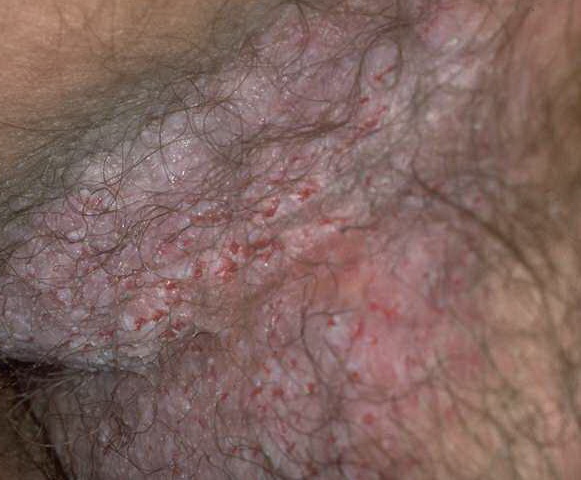
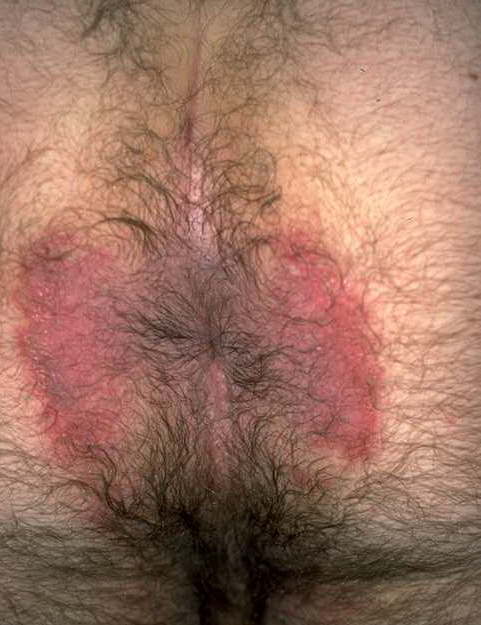
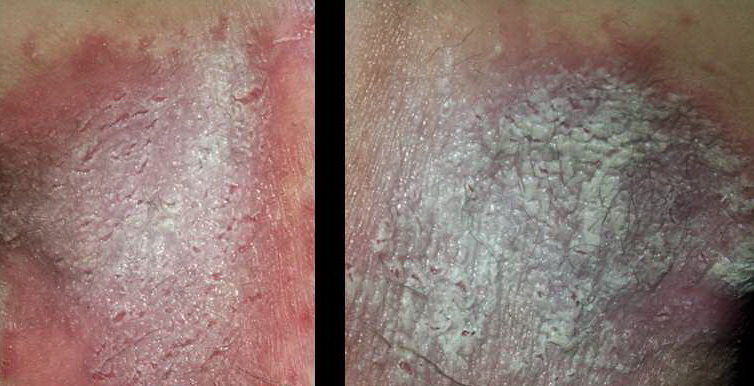
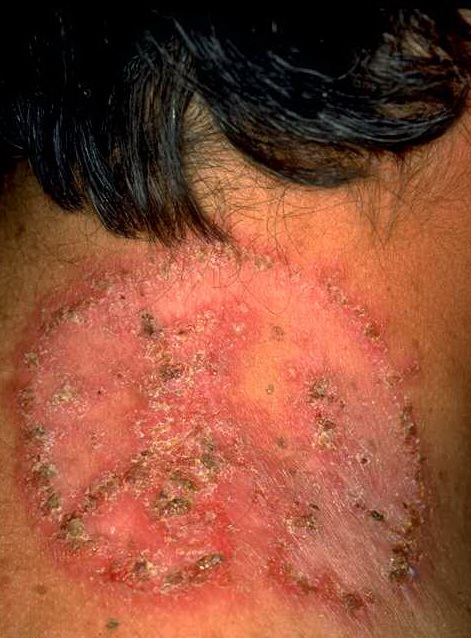
With familial benign pemphigus, vesicles and erythematous plaques with overlying crusts typically occur in the genital area, as well as the chest, neck, and axillary areas . Burning and itching accompany the eruption, and a malodorous drainage occurs in some cases as a result of secondary infection. Symptoms related to staphylococcal and candidal overgrowth are common in familial benign pemphigus. Multiple asymptomatic longitudinal white bands on the fingernails also have been described. Involvement of mucosa is rare. The characteristic clinical appearance of familial benign pemphigus, as well as biopsy findings, readily confirms the diagnosis.
Hailey-Hailey disease, or familial benign pemphigus, is hypothesized to result from a genetic defect in a calcium pump protein. The pump mutation is in ATP2C1, a gene localized on chromosome 3. This gene defect is similar to the genetic defect in Darier disease, which also is a calcium pump defect, ATP2A2. The gene ATP2C1 encodes the human secretory pathway Ca++ -ATPase hSPCA1, which is dysfunctional and causes abnormal calcium release from the Golgi apparatus and endoplasmic reticulum.
In addition to the primary gene defect in Hailey-Hailey disease (familial benign pemphigus), contributing factors are known that exacerbate the disease. These include heat, friction, and infection, resulting in separation of keratinocytes, especially in the intertriginous areas. Through ultrastructural studies of familial benign pemphigus lesions, characteristic changes in keratinocyte morphology have been described, including retracted tonofilaments, elongated membrane microvilli, and reduced numbers of desmosomes.