| Parapsoriasis Large-plaque = نظير الصدف كبير اللويحات |
|
|
Parapsoriasis
Historical Aspects the currently accepted classification of parapsoriasis includes large and small-plaque forms of parapsoriasis en plaques (often referred to simply as parapsoriasis) as well as acute and chronic forms of pityriasis lichenoides [known today as pityriasis lichenoides et varioliformis acuta (PLEVA) and pityriasis lichenoides chronica (PLC), respectively].1 Pityriasis lichenoides was first described in 1894 by Neisser7 and Jadassohn.8 In 1899 Juliusberg delineated the chronic form and named it PLC.Mucha redescribed the acute form in 1916 and distinguished it from the chronic form. Habermann named the acute variant PLEVA in 1925. Mucha-Habermann disease is synonymous with PLEVA. Some authors regard lymphomatoid papulosis as a variant of pityriasis lichenoides, whereas others consider it to be a separate disease. Lymphomatoid papulosis is discussed in Chap. 146 as part of the spectrum of CD30+ cutaneous lymphoproliferative disorders.
Epidemiology Large-plaque parapsoriasis (LPP) and small-plaque parapsoriasis (SPP) are, in general, diseases of middle-aged and older people, with a peak incidence in the fifth decade. Occasionally, lesions arise in childhood and may be associated with pityriasis lichenoides. SPP shows a definite male predominance of approximately 3:1. LPP is probably more common in males, but the difference is not as striking as in SPP. Both occur in all racial groups and geographic regions. Etiology and Pathogenesis It is likely that a complete understanding of the pathogenesis of parapsoriasis will develop with our understanding of the pathogenesis of both chronic dermatitis and mycosis fungoides (MF), because parapsoriasis appears to bridge these disorders. The T cells that mediate most inflammatory skin diseases belong to the skin-associated lymphoid tissue (SALT).15 These T cells express the cutaneous lymphocyte-associated antigen and traffic between the skin and the T-cell domains of peripheral lymph nodes via the lymphatics and bloodstream. MF has been shown to be a neoplasm of SALT T cells. Sensitive polymerase chain reaction (PCR)-based tumor clonality assays have underscored the SALT nature of MF tumor clones by showing that they can continue to traffic after neoplastic transformation16 and can even participate in delayed-type hypersensitivity reactions to contact allergens.17 This implies that rather than being a skin lymphoma per se, MF is actually a SALT lymphoma; that is, a malignancy of a T-cell circuit rather than of one particular tissue. Trafficking of MF tumor cells has been detected even in patients with very early stage disease whose lesions were consistent clinicopathologically with LPP.16,18 Therefore, it can be said that at least in some cases LPP is a monoclonal proliferation of SALT T cells that have the capacity to traffic between the skin and extracutaneous sites. This view is also supported by the presence of structural and numerical chromosomal abnormalities in the peripheral blood mononuclear cells of patients
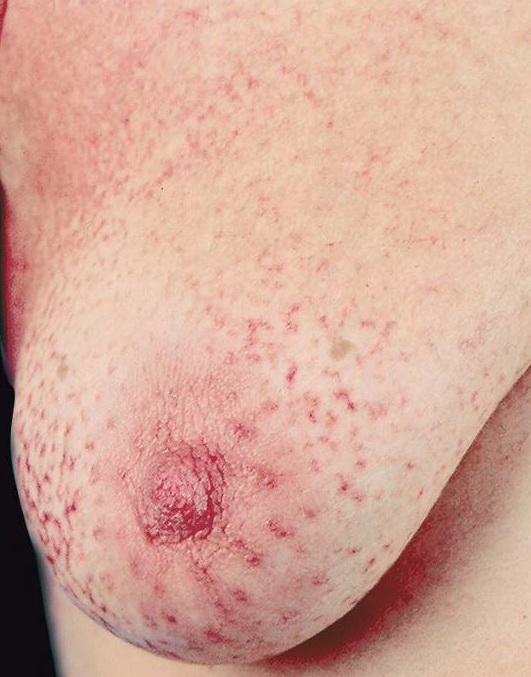
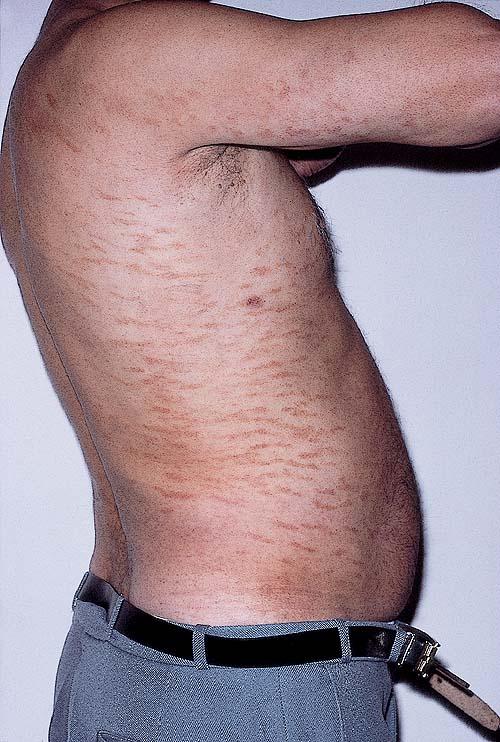
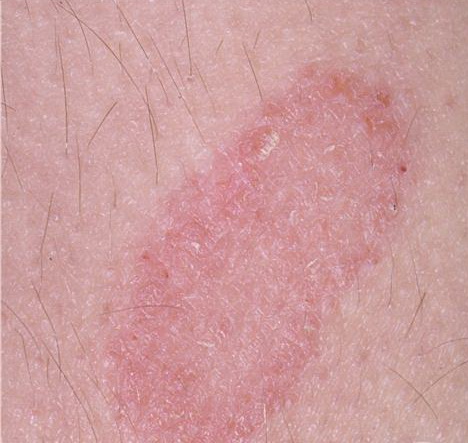
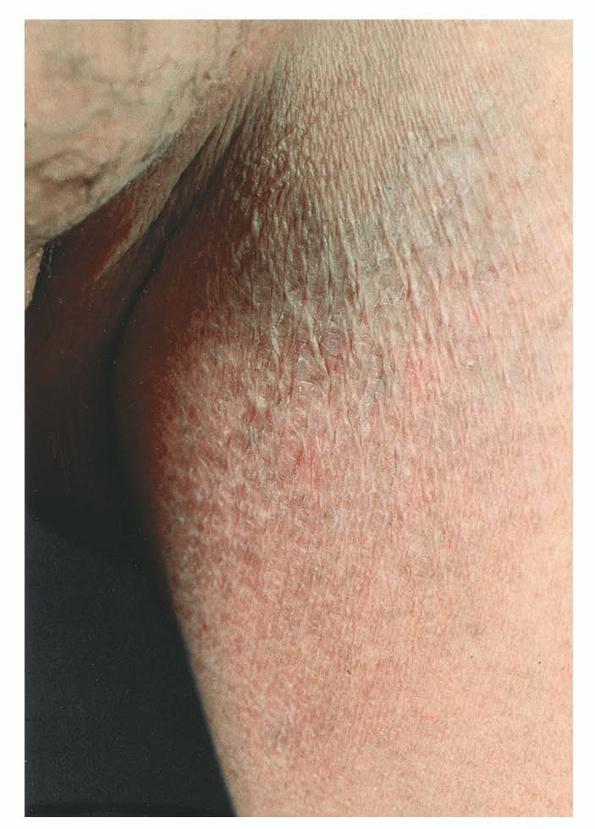
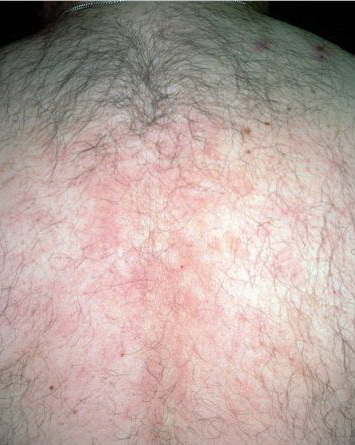
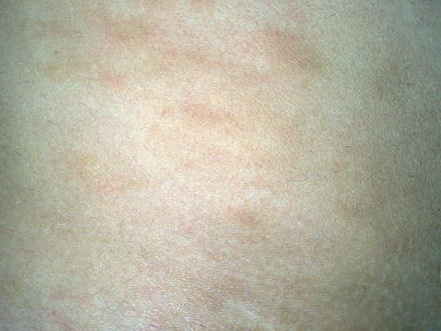
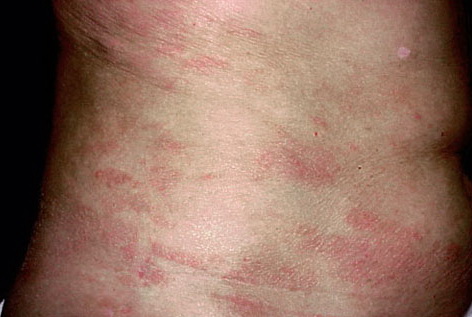
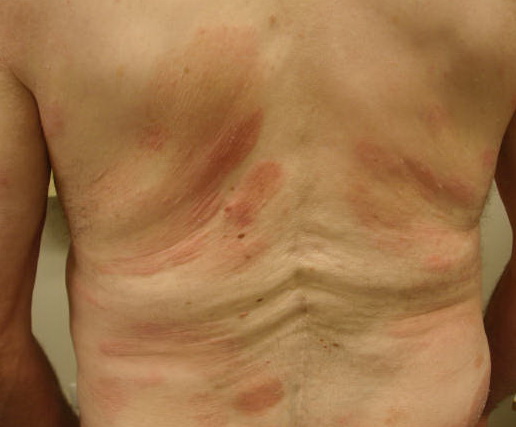
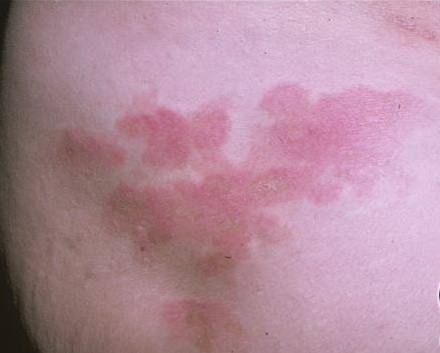
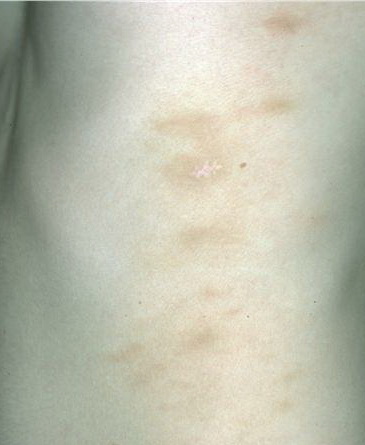
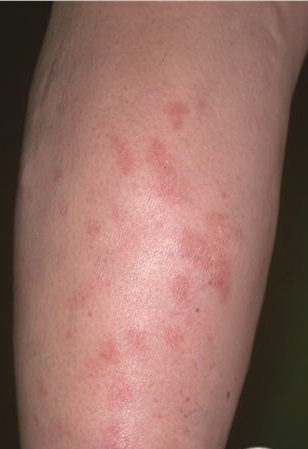
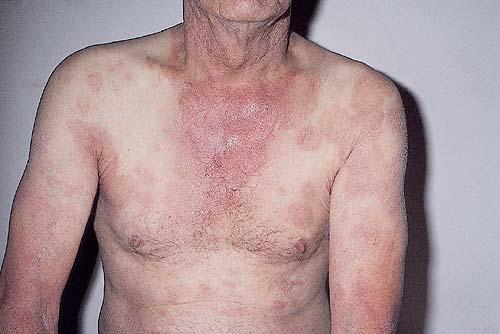
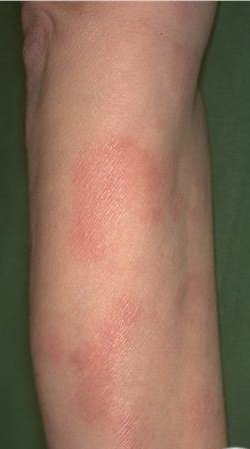
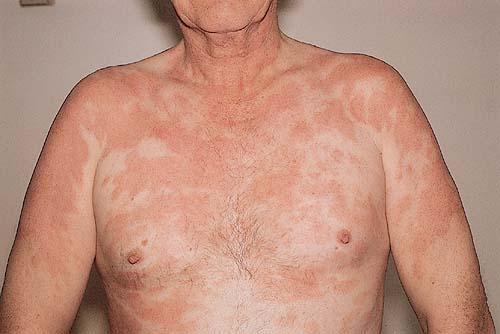
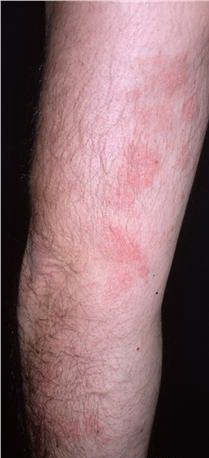
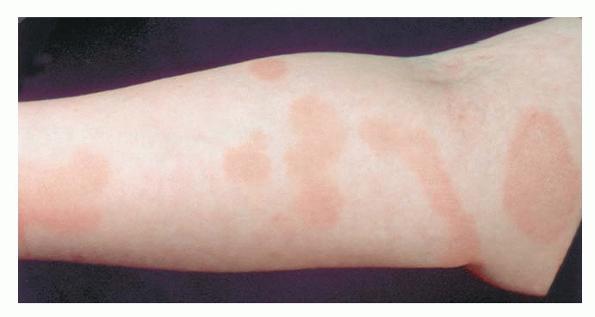
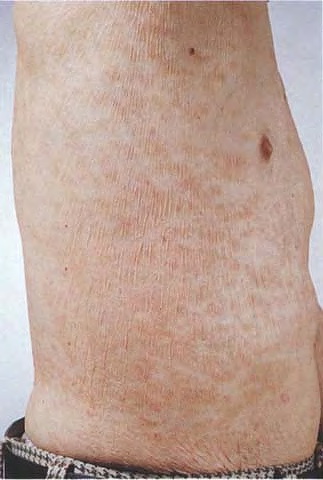
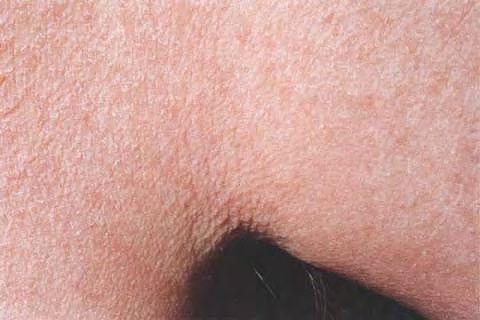
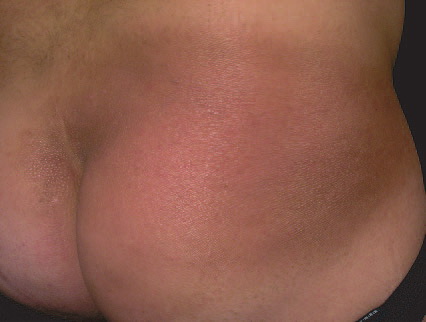
A unifying feature of the parapsoriasis group of diseases is that all of them appear to be cutaneous T-cell lymphoproliferative disorders: LPP, SPP, pityriasis lichenoides,33,35-37 and lymphomatoid papulosis have all been shown to be monoclonal disorders in many cases.5 These relationships suggest that progression from LPP through the various stages of the MF disease spectrum is accompanied by an increasing gradient of dominant T-cell clonal density resulting from mutations that confer increasing growth autonomy to the neoplastic T-cell clone. Interestingly, analysis of peripheral blood has demonstrated that clonal T cells are often detectable in patients with LPP/early MF31,33 or SPP, which again supports the systemic SALT nature of these “primary” skin disorders. Dominant clonality as seen in the parapsoriasis disease group, follicular mucinosis, pagetoid reticulosis, and certain other disorders does not equate to clinical malignancy. In fact, most patients with these diseases experience a benign clinical course, and in some cases the disease resolves completely. In addition, other types of chronic cutaneous T-cell infiltrates sometimes exhibit dominant clonality, including primary (idiopathic) erythroderma and non-specific chronic spongiotic dermatitis. This has given rise to the concept of clonal dermatitis, originally described in the context of clonal non-specific chronic spongiotic dermatitis but later expanded to include other nonlymphomatous cutaneous T-cell infiltrates that harbor occult monoclonal T-cell populations. Several cases of clonal dermatitis, some of which have progressed to MF, have been identified.18,28,32 We suspect that for each disease with a potential for progression to MF, the principal risk may reside in the subset showing clonal dermatitis, because this is the subset in which dysregulation has begun to occur. Clinical Findings CUTANEOUS LESIONS LPP lesions are either oval or irregularly shaped patches or very thin plaques that are asymptomatic or mildly pruritic. They are usually well marginated but may also blend imperceptibly into the surrounding skin. The size is variable, but typically most lesions are larger than 5 cm, often measuring more than 10 cm in diameter. Lesions are stable in size and may increase in number gradually. They are found mainly on the “bathing trunk” and flexural areas . Extremities and the upper trunk, especially the breasts in women, also may be involved. They are light red-brown or salmon pink, and their surface is covered with small and scanty scales. Lesions may appear finely wrinkled—“cigarette paper” wrinkling. Such lesions exhibit varying degrees of epidermal atrophy. Telangiectasia and mottled pigmentation also are observed when the atrophy becomes prominent (Fig. 25-3). This triad of atrophy, mottled pigmentation, and telangiectasia defines the term poikiloderma or poikiloderma atrophicans vasculare, which also may be seen in other conditions . Retiform parapsoriasis refers to a rare variant of LPP that presents as an extensive eruption of scaly macules and papules in a net-like or zebra-stripe pattern that eventually becomes poikilodermatous . SPP characteristically occurs as round or oval discrete patches or very thin plaques, mainly on the trunk . The lesions measure less than 5 cm in diameter; they are asymptomatic and covered with fine, moderately adherent scales. The general health of the patient is unaffected. A distinctive variant with lesions of a finger shape, known as digitate dermatosis, has yellowish or fawn-colored lesions . It follows lines of cleavage of the skin and gives the appearance of a hug that left fingerprints on the trunk. The long axis of these lesions often measures greater than 5 cm. Chronic superficial dermatitis is a synonym for SPP.43 Digitate lesions with a yellow hue were referred to in the past as xanthoerythrodermia perstans. Differential Diagnosis of Poikiloderma
Laboratory Tests
HISTOPATHOLOGY In early LPP lesions, the epidermis is mildly acanthotic and slightly hyperkeratotic with spotty parakeratosis. The dermal lymphocytic infiltrate tends to be perivascular and scattered . In the more advanced lesions one observes an interface infiltrate with definite epidermotropism. These invading lymphocytes may be scattered singly or in groups, sometimes associated with mild spongiosis. In addition, the poikilodermatous lesions show atrophic epidermis, dilated blood vessels, and melanophages . Immunohistologic studies have revealed similar features in LPP and early MF, including a predominance of CD4+ T-cell subsets, frequent CD7 antigen deficiency, and widespread epidermal expression of class II HLA (HLA-DR). SPP exhibits mild spongiotic dermatitis with focal areas of hyperkeratosis, parakeratosis, scale crust, and exocytosis. In the dermis, there is a mild superficial perivascular lymphohistiocytic infiltrate and dermal edema . There is no progression of the histologic features with time. Immunohistologic studies reveal a predominantly CD4+ T-cell infiltrate with non-specific features resembling those seen in various types of dermatitides. Diagnosis and Differential Diagnosis LPP is distinguished from SPP by the larger size, asymmetric distribution, and irregular shape of its lesions, which are
The degree to which LPP is differentiated from early MF depends primarily on the histopathologic criteria used to diagnose the latter disorder. Unfortunately, there are no universally accepted minimal criteria for the diagnosis of MF; however, one set proposed by the International Society for Cutaneous Lymphoma is presented in Table 25-2. This algorithm is based on a holistic integration of clinical, histopathologic, immunopathologic, and clonality data. It differs significantly from many prior approaches because it does not rely solely on histopathologic features.50 Assuming that histopathologic examination does not disclose features diagnostic of some other dermatosis, these criteria allow lesions to be classified as either patch-stage MF or not. For the practical purposes of clinical management, patients presenting clinically with patch lesions whose features result in a score of 4 points or more are considered to have unequivocal MF. Obviously, the more liberal the criteria, the more cases could be considered to be MF. However, there will always be some cases that fail to meet any specific set of criteria, and the designation LPP is a useful term to apply to them because it guides treatment and follow-up and conveys an understanding that the risk of dying from lymphoma is small.
The clinical and/or histopathologic differential diagnosis of LPP also includes those collagen vascular diseases and genodermatoses exhibiting poikilodermatous features, lichenoid drug eruptions, secondary syphilis, chronic radiodermatitis, and occasionally several other diseases tabulated in Box 25-2. These generally can be distinguished by their associated clinical findings. Histopathologic differentiation among these diseases is covered largely in the discussion of pseudo-MF in Chap. 147. SPP, when it presents with its distinctive digitate dermatosis lesions parallel to skin lines in a truncal distribution, stands out from other types of parapsoriasis. Individual SPP lesions may show some superficial resemblance to PLC. SPP is distinguished from psoriasis by the absence of the Auspitz sign , micaceous scale, nail pits, and typical psoriatic lesions involving the scalp, elbows, and knees. Histologically, its mild spongiotic dermatitis and absence of other characteristic features distinguish it from PLC, psoriasis, and several of the other entities listed in Box 25-2. Clinical features are also important, such as the herald patch of pityriasis rosea and the papulovesicular coin-shaped patches favoring the lower extremities in nummular dermatitis. Sometimes patients with MF may exhibit small patches of disease at presentation; however, these lesions typically have histopathologic features at least consistent with MF and generally are associated with larger, more classic lesions of MF elsewhere on the skin. They also may show poikilodermatous features not seen in SPP. Furthermore, the presence of well-developed, moderate to thick small plaques, as seen in some MF patients, is incompatible with the diagnosis of SPP because the latter disorder includes only lesions that are no more than patches or very thin plaques. It is also important to recognize that partially treated or early relapsing lesions of MF may show only non-specific features that should not be taken as evidence of a pathogenetic link to SPP or any other dermatosis. Complications LPP can be associated with other forms of parapsoriasis and overt cutaneous
Differential Diagnosis of Large-Plaque Parapsoriasis (LPP) and Small-Plaque Parapsoriasis (SPP) Most Likely
Consider
Always Rule Out
Prognosis and Clinical Course Both LPP and SPP may persist for years to decades with little change in appearance clinically or histopathologically. Approximately 10 to 30 percent of cases of LPP progress to overt MF. In this context, LPP represents the clinically benign end of the MF disease spectrum, with transformation to large cell lymphoma at the opposite extreme. The rare retiform variant is said to progress to overt MF in virtually all cases.
In contrast to LPP with its malignant potential, SPP is a clinically benign disorder in the experience of most experts. Patients with this disease as defined in this chapter rarely, if ever, develop overt MF.15,43,52 Despite this fact and what most observers consider to be its nonspecific histopathologic features, some authors favor lumping SPP within the MF disease spectrum as a very early, nonprogressive variant.53 This issue has been debated at length.36,54,55 Treatment of Large-Plaque Parapsoriasis and Small-Plaque Parapsoriasis FIRST LINE
SECOND LINE (Mainly for large-plaque parapsoriasis cases considered to be early mycosis fungoides)
Treatment Patients with SPP should be reassured and may forego treatment. The disease may be treated with emollients, topical tar preparations, topical corticosteroids, and/or broadband or narrowband ultraviolet B (UVB) phototherapy . Response to therapy is variable. Patients should initially be examined every 3 to 6 months and subsequently every year to ensure that the character of the process is stable. LPP requires more aggressive therapy: high-potency topical corticosteroids with phototherapy such as broadband UVB, narrowband UVB, or psoralen and ultraviolet A (PUVA). The goal of treatment is to suppress the disorder to prevent possible progression to overt MF. Other methods of treatment, such as topical nitrogen mustard, have been used, particularly for the poikilodermatous type. The patient should be examined carefully every 3 months initially and every 6 months to 1 year subsequently for evidence of progression. Repeated multiple biopsies of suspicious lesions should be performed. Cases that satisfy the clinicopathologic criteria for early MF can be treated with broadband UVB, narrowband UVB, PUVA, topical nitrogen mustard, topical bexarotene gel, topical imiquimod, or topical carmustine (BCNU). Electron-beam radiation therapy generally is reserved for more advanced, infiltrated lesions of MF. Prevention Because LPP and SPP are uncommon diseases that appear to affect patients randomly, there are no known preventive measures.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||